Severe or profound sensorineural hearing impairment (SNHI) is a common birth defect, affecting approximately 1 in 1000 newborns [1]. SNHI may result from environmental causes or have a genetic basis. The genetic causes can be further subdivided into non-syndromic (isolated HI) or syndromic, where the HI forms part of a more complex medical disorder.
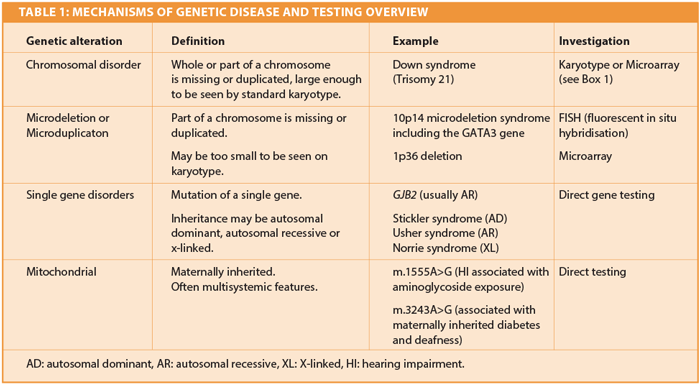
The human genome is encoded as DNA sequences packaged into the 23 pairs of chromosomes and mitochondrial DNA. A genetic disorder can be defined as a condition caused by a pathological alteration in the genome. Variation in the genome is normal and usually not detrimental. Some alterations do lead to clinical problems, usually as a result of abnormal expression of the protein(s) encoded by the affected gene(s). This can in turn cause abnormal growth and development and sometimes the outcome is a recognisable genetic disorder. A genetic disorder may be inherited or occur sporadically, due to new mutations. Genetic alterations can occur at several levels, summarised in Table 1.
Non-syndromic hearing impairment
There are over 100 known loci for non-syndromic SNHI and more remain to be discovered. This includes autosomal dominant, autosomal recessive and X-linked variants [2]. Mutation in GJB2 (encoding the protein connexin 26) is by far the most common genetic cause of non-syndromic SNHI and accounts for up to 50% of recessive cases in some populations [3]. GJB2 associated SNHI is usually congenital and is frequently detected by the Newborn Hearing Screening programme. It is most commonly seen as an autosomal recessive trait, although a few specific, rare mutations may cause dominantly inherited hearing impairment, which may be either syndromic or non-syndromic.
Syndromic hearing impairment
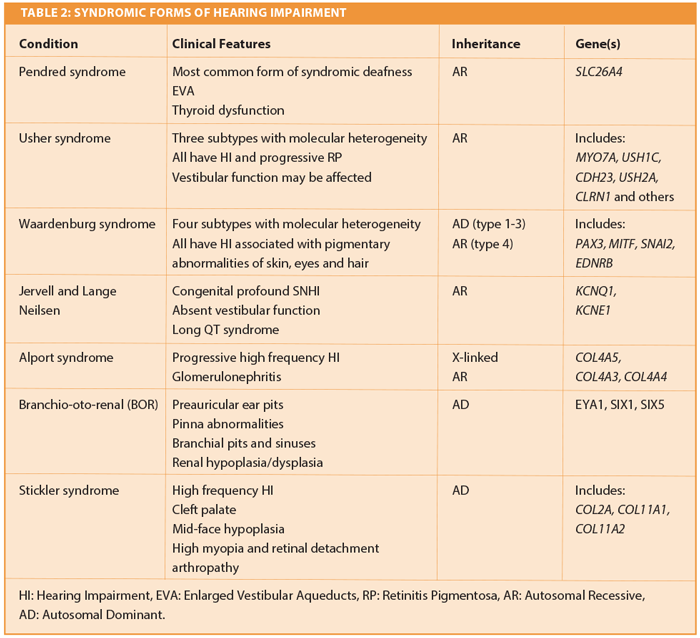
Syndromic HI accounts for approximately 30% of genetic SNHI. There are several hundred (mostly rare) reported syndromes associated with SNHI; some of the more common or diagnostically important ones are summarised in Table 2 [4]. Establishing a syndromic genetic diagnosis is important for several reasons:
- There may be surveillance implications to consider such as monitoring for thyroid dysfunction in Pendred syndrome, or renal dysfunction in branchio-oto-renal syndrome
- It may inform management decisions regarding the benefit or otherwise of cochlear implant. Patients with Usher syndrome for example may lose vision and therefore lose the ability to communicate by sign language, so early bilateral cochlear implantation is important
- It may suggest other preventative measures such as the avoidance of aminoglycosides in some cases of mitochondrial HI
- It allows accurate genetic counselling and estimation of recurrence risk in a family, which is important to parents.
Assessment of a child presenting to genetics with hearing loss
History and examination
As with all specialities, the assessment of a patient in the genetics service consists of taking a good history and examination. In addition, drawing a three generation pedigree and enquiring specifically about consanguinity and ethnicity are important. A history of consanguinity may suggest recessive inheritance and homozygosity for a particular mutation. Some mutations are particularly prevalent in certain ethnic groups and may be screened for quickly. Other relevant questions include antenatal and birth history, developmental milestones (delayed walking is often seen with vestibular involvement), personal or family history of renal disease (Alport syndrome and Branchio-oto-renal syndrome) or ophthalmological involvement (Usher syndrome, Stickler syndrome and Norrie disease). A full examination should be performed including a detailed inspection of the ears, skin and associated structures.
Investigations
These should be tailored to the history; for a good review see [5]. Often non-genetic investigations may give clues as to an underlying aetiology and may help direct subsequent genetic tests. These include:
- Audiograms in patient and parents. These may indicate an inheritance pattern or be typical for a particular disorder (‘ski-slope’ audiogram appearance in TMPRSS3 associated HI, or low frequency non-syndromic HI seen as a result of mutation of WFS1). They should be sent with referral where possible
- CMV testing in order to exclude a common environmental cause of HI
- Imaging of the inner ear (enlarged vestibular aqueducts in Pendred syndrome, absent semicircular canals in CHARGE syndrome)
- Urinalysis for haematuria in Alport syndrome
- Ophthalmology review (in all cases to correct refractive error but also to examine for retinitis pigmentosa in established Usher syndrome and retinal detachment in Norrie disease)
- Vestibular assessment if motor delay is a feature. Confirmed vestibular involvement should prompt:
º ECG (Jervell and Lange-Nielsen syndrome)
º Electroretinogram (Usher syndrome)
º MRI / CT scan of the inner ear.
Genetic investigations
In order to arrange the appropriate investigations a basic knowledge of the different types of genetic disorder and associated testing is required (Table 1).
In practice, an individual presenting with non-syndromic congenital or childhood-onset HI will usually have GJB2 screened at an early stage. If there are additional features that clearly point to a specific cause, targeted gene testing may be appropriate (e.g. pigmentary abnormalities in Waardenburg syndrome). A child with HI and dysmorphic features may have a microarray to exclude a microduplication or microdeletion (Box 1).

The last few years have seen huge advances in genetic technology and have paved the way for new diagnostic approaches in clinical genetics. These are now starting to filter through into clinical practice and have the potential to hugely improve diagnostic rates. Of particular note is the development of specialised gene panel tests and the advent of exome sequencing (Box 2). Exome sequencing refers to sequencing of all of the protein-coding ‘exons’ in the genome. Although not yet widely available on a routine basis, both are increasingly forming part of clinical genetic practice.
Conclusion
Consideration of a genetic aetiology should form part of the routine evaluation of an individual with HI, with or without a family history and can positively impact management strategies for the patient and other family members. Referral to clinical genetics service may be beneficial in cases where there is a family history of HI or where a syndromic cause is likely.
References
1. Fortnum HM, Summerfield AQ, Marshall DH, et al. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: questionnaire based ascertainment study. BMJ 2001;323(7312):536-40.
2. Hereditary hearing loss.
http://hereditaryhearingloss.org
Last accessed September 2014.
3. Gasparini P, Rabionet R, Barbujani G, et al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur J Hum Genet 2000;8(1):19-23.
4. London Medical Database.
http://suite.face2gene.com/lmd-history/
Last accessed September 2014.
5. Ardle BM, Bitner-Glindzicz M. Investigation of the child with permanent hearing impairment. Arch Dis Child Educ Pract Ed 2010;95(1):14-23.
6. Shearer AE, Black-Ziegelbein EA, Hildebrand MS, et al. Advancing genetic testing for deafness with genomic technology. J Med Genet 2013;50(9):627-34.
7. Diaz-Horta O, Duman D, Foster J 2nd, et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS One 2012;7(11):e50628.
Declaration of competing interests: None declared.



