Otitis media (OM), a common disease of childhood, is considered to be a complex trait with multiple genetic and environmental factors expected to contribute to a child’s risk of developing recurrent acute OM (rAOM; ≥3 episodes in 6 months or ≥4 episodes in a year) or chronic OM with effusion (COME; persistent middle ear fluid for ≥3 months).
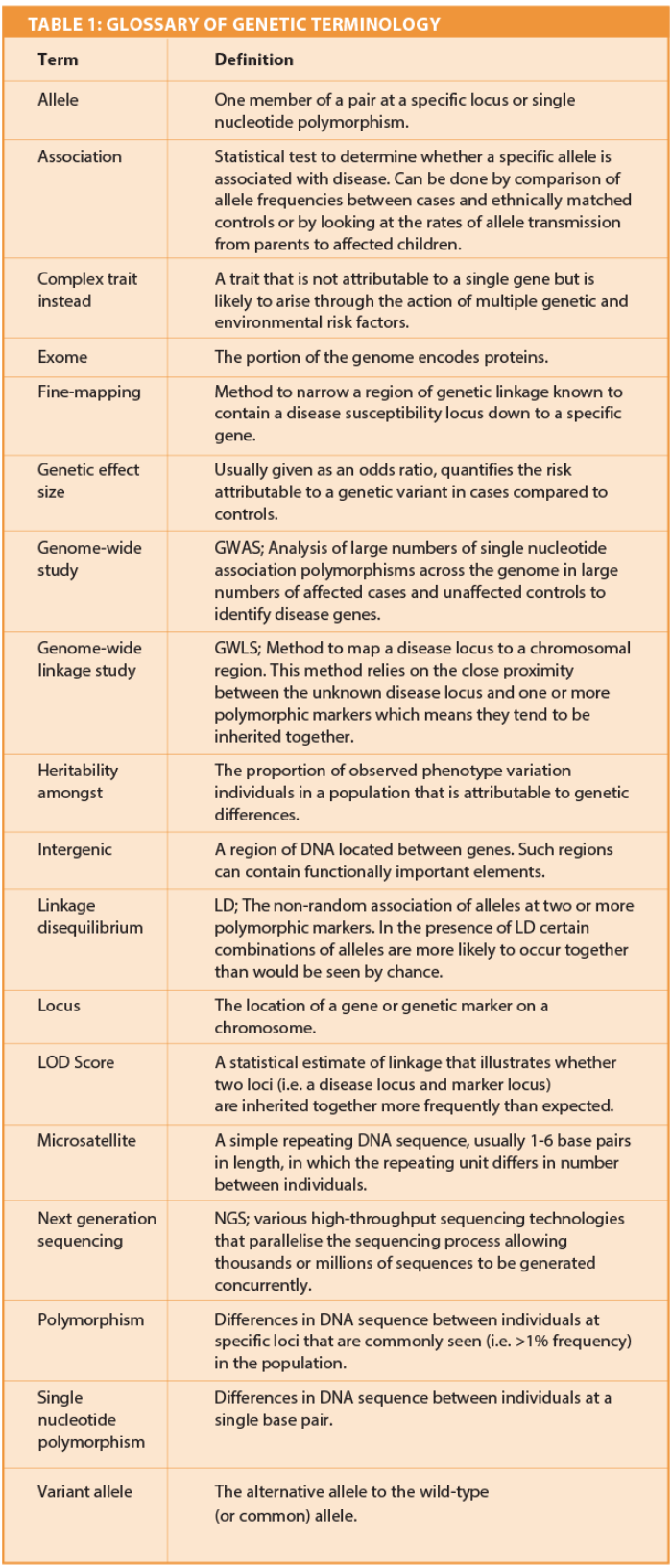
Heritability studies confirm the genetic component is likely to be substantial, explaining 40-70% of the variation in OM phenotype [1]. In order to identify the genes involved in susceptibility to OM several study designs have been employed to date and are described briefly in this article (Table 1).
The most commonly used approach has been the candidate gene association study. This study design investigates the alleles observed at a small number (10-100) of single nucleotide polymorphisms (SNPs) located at or close to specific genes, either in a cohort of individuals with disease (cases) compared to those without disease (controls) or within families (usually parent/child trios). The specific genes investigated are usually selected on the basis of plausible biological function or due to their homology with disease genes identified in relevant murine models. Statistical evidence that specific SNP alleles differ in frequency between cases and controls suggests that the SNP either directly increases risk of the disease (i.e. it is a causal variant) or that it is in linkage disequilibrium (LD) with the true causal variant.
The alternative approach has been to interrogate the whole genome using either a genome-wide linkage study (GWLS) or a genome-wide association study (GWAS) design. In a GWLS hundreds of microsatellite markers or thousands of SNPs spaced evenly across the genome are genotyped in sets of affected relatives to identify regions that are co-inherited due to genetic linkage. The linked genomic regions identified tend to be large (often megabases in size) and frequently harbour tens to hundreds of genes, any one of which could ultimately be the disease locus. As such, results from a GWLS have to be followed up by fine-mapping of all genes under the linkage peak, a task usually achieved via an association study.
“The most commonly used approach has been the candidate gene association study.”
More recently the GWAS approach has been utilised to identify the genes involved in susceptibility to OM. In a GWAS the frequencies of (usually) several million SNPs are compared across the whole genome in hundreds or thousands of unrelated cases versus ethnically matched controls. Again, statistical evidence that specific SNP alleles differ in frequency between cases and controls suggests a role for those SNPs, or ones they are in LD with, in disease. The GWAS has become a popular approach for the investigation of many complex diseases as previously unsuspected genetic risk factors can be identified which may provide novel insight into gene networks / pathways important in disease pathogenesis. In the context of OM the GWAS approach has only recently been undertaken for several reasons, the key one of which is the availability of large case/control cohorts. Such large case/control sample sizes are essential to detect the small genetic effect sizes (i.e. Odds Ratio [OR] ≤1.5) expected to contribute to a complex trait such as OM.
Regardless of the study design chosen, it is essential that replication studies are subsequently performed to reduce the number of false positive genetic risk factors reported. Replication is ideally performed in an independent case/control sample drawn from the same population that uses a comparable phenotype and which is of sufficient size to replicate the genetic effect size observed in the original analysis. Without replication, results should be interpreted with some caution.
Our understanding of the genetic risk factors that are important in childhood OM has advanced considerably in recent years. The results of numerous candidate gene studies have been reported across several different populations and have identified positive association at more than 20 individual genes (recently reviewed [2]). This includes several genes involved in the immune response, such as Toll-like Receptor 4 (TLR4), members of the Interleukin (IL) family of cytokines (IL1A, IL1B, IL6, IL10), Tumour Necrosis Factor alpha (TNF) and several members of the Mucin gene family (MUC2, MUC5AC, MUC5B). However, it is important to note that many of these reported candidate gene associations have been based on small sample sizes and very few have been replicated in independent populations to date.
One notable exception is the association reported at the F-Box only Protein 11 (FBXO11), a gene initially identified in a murine model of OM [3]. Association has been observed at variants in this gene in four independent (predominantly Caucasian) cohorts from Australia, the USA and the UK making this one of the strongest genetic risk factors for OM reported to date [4]. Subsequent functional work in the Fbxo11 mouse model has highlighted the role of middle ear hypoxia in OM and points to molecules that target hypoxia-mediated pathways as potential novel therapies to alleviate middle ear hypoxia, accumulation of middle ear fluid and the associated conductive hearing loss [5].
Several genome-wide studies investigating OM genetic risk factors have also been reported. This includes two independent GWLS that have identified several regions of genetic linkage on chromosomes 3p25, 10q22, 10q26, 17q12 and 19q13, all of which appear to contain disease susceptibility loci [6, 7]. To date one independent replication study has been reported which confirmed the genetic linkage reported at chromosome 10q26 only. Subsequent fine mapping of the 10q26 region suggests a role for the Protein Phosphatase 2, Regulatory Subunit B, Delta (PPP2R2D) gene, a modulator of the Transforming Growth Factor ß (TGF-ß) pathway [8]. The remaining linkage regions have yet to be replicated and the causative disease genes contained within them identified.
More recently the results from the first GWAS of OM have been reported. The first GWAS of OM identified associations at the Calpain 14 (CAPN14) and N-acetylgalactosaminyltransferase 14 (GALNT14) genes on chromosome 2p23.1, as well as association at two members of the Bacterial Permeability Increasing Protein Family (BPIFA3 and BPIFA1) cluster on chromosome 20q11.21 [9]. These gene products play a role in the invasion, adherence or recognition of respiratory bacterial pathogens respectively, pointing to the potential functional significance of these novel genetic risk factors. The second GWAS of OM identified associations at several different genes/regions, including at the Kinesin Family Member 7 (KIF7) gene and a SNP within an intergenic region on chromosome 2q31.1 that has been shown to regulate the Low-Density Lipoprotein Receptor (LDLR) expressed on ciliated airway epithelial cells [10]. To date, the association at the 2q31.1 intergenic SNP is the only GWAS association to have been replicated in an independent cohort.
Whilst it is known that microbial and environmental factors play an important role in OM there is also a clear role for genetic factors, one that we are only just starting to understand. The recent formation of OTIGEN, an international consortium of researchers focused on delineating the genetic basis of OM, will greatly facilitate ongoing investigation of the genetic risk factors by ensuring the availability of large, precisely phenotyped cohorts for initial analyses and subsequent replication studies. Rapidly advancing genomic technologies will also contribute to ongoing investigations. Studies undertaken so far have focused on the role played by common polymorphism. In the future it is likely that attention will also turn to the role played by rare polymorphism/mutation.
Recent advances made in next generation sequencing (NGS) methods now provide the means with which to sequence the entire human exome and rapidly falling costs mean that it will soon be possible to undertake such analyses in large numbers of samples. Overall, the identification of the genes that contribute to OM susceptibility will provide important insights into the biological complexity of this disease and could ultimately contribute to improved preventative and therapeutic strategies to reduce disease incidence.
References
1. Casselbrant ML, Mandel EM, Fall PA, et al. The heritability of otitis media: a twin and triplet study. JAMA 1999;282:2125-30.
2. Rye MS, Blackwell JM, Jamieson SE. Genetic susceptibility to otitis media in childhood. The Laryngoscope 2012;122:665-75.
3. Hardisty-Hughes RE, Tateossian H, Morse SA, et al. A mutation in the F-box gene, Fbxo11, causes otitis media in the Jeff mouse. Human Molecular Genetics 2006;15:3273-9.
4. Rye MS, Bhutta MF, Cheeseman MT, et al. Unraveling the genetics of otitis media: from mouse to human and back again. Mammalian Genome: Official J Int Mammalian Genome Society 2011;22:66-82.
5. Cheeseman MT, Tyrer HE, Williams D, et al. HIF-VEGF pathways are critical for chronic otitis media in Junbo and Jeff mouse mutants. PLoS Genetics 2011;7:e1002336.
6. Casselbrant ML, Mandel EM, Jung J, et al. Otitis media: a genome-wide linkage scan with evidence of susceptibility loci within the 17q12 and 10q22.3 regions. BMC Medical Genetics 2009;10:85.
7. Daly KA, Brown WM, Segade F, et al. Chronic and recurrent otitis media: a genome scan for susceptibility loci. American J of Human Genetics 2004;75:988-97.
8. Rye MS, Scaman ES, Thornton RB, et al. Genetic and functional evidence for a locus controlling otitis media at chromosome 10q26.3. BMC Medical Genetics 2014;15:18.
9. Rye MS, Warrington NM, Scaman ES, et al. Genome-wide association study to identify the genetic determinants of otitis media susceptibility in childhood. PloS One 2012;7:e48215.
10. Allen EK, Chen WM, Weeks DE, et al. A genome-wide association study of chronic otitis media with effusion and recurrent otitis media identifies a novel susceptibility locus on chromosome 2. J Assoc Res Otolaryngol 2013;14:791-800.
Declaration of competing interests: None declared.


