By their very nature, late-onset hereditary disorders offer a large window of opportunity for therapeutic intervention. However, before we can begin to think about strategies we need knowledge of the genetics and pathology underlying the condition. In this article we discuss the utility of the mouse as a model organism for gene discovery and validation in late-onset hereditary diseases, using age-related hearing loss (ARHL) as an example.
Introduction
ARHL, also known as presbyacusis, is a progressive bilateral symmetrical age-related sensorineural hearing loss, which is most pronounced at the higher frequencies. It is a significant health burden on the population, being the most prevalent sensory impairment observed in the elderly and one of the four most common chronic health conditions experienced in old age. ARHL not only leads to communication difficulties, it is also associated with social isolation, depression, and reduced physical and cognitive function [1].
It is a complex condition, with contribution from both environmental (e.g. noise, ototoxic drugs) and genetic factors. Indeed, twin and family studies have shown high heritability (~50%), suggesting that genetics has a fundamental role in susceptibility to this condition [2-4]. As for many complex disorders genome-wide association (GWA) studies have been undertaken in an effort to elaborate upon the genes involved in adult auditory function [5-9]. However, in the several studies published to date, no genes demonstrate genome-wide significance, but that is not to say the genes showing some degree of association will not prove to be involved in ARHL.
Regarding the pathology of ARHL, Schuknecht proposed four pathological subtypes: sensory, involving hair cell loss; strial (or metabolic), involving degeneration of the stria vascularis and reduction in endocochlear potential; neural, involving loss of spiral ganglion neurons; and mechanical, involving stiffening of the basement membrane, although the latter is now thought to be a manifestation of strial presbycusis [10-12]. The study of disease onset and progression is hampered by the lack of access to tissue specimens, but it is likely most people with ARHL have a mixed pathology.
Given the difficulties involved with studying the genetics and pathological progression of ARHL in humans (e.g. long lifespan, accumulated environmental influences, genetic heterogeneity, and ethical issues), researchers have employed animal models.
Mouse as a model for hearing research
Mice continue to be the predominant model organism for hearing research. The reasons for this are many-fold and include: similarities in auditory structure and physiology between mice and humans; the close evolutionary relationship of genomes (~99% of genes in mice have a human orthologue); and, relatively low housing costs (small size, short lifespan). However, arguably the most important reason is the available genetic toolkit we have at our disposal for manipulating the murine genome [13]. In addition, the mouse is ideal for ageing research as it has a short lifespan, which means the effects of ageing become apparent within a contracted time period and tissues can be obtained for analysis at all stages of the disease process. Other factors that make the mouse a suitable model are the ability to control their environment (e.g. diet, noise exposure, etc) and genetic background.
As well as spontaneous mutations that cause hearing loss in mice, there are a variety of tools available to manipulate the mouse genome and identify genes involved with both early and late-onset deafness. The approaches available can be loosely divided into phenotype-driven approaches and gene-driven approaches.

For phenotype-driven approaches, we randomly mutagenize the mouse genome with a chemical mutagen, ENU (N-ethyl-N-nitrosourea), and then screen mice for hearing loss. We discuss this approach below in relation to screens for genes involved with presbyacusis. The advantage of phenotype-driven screens is that they make no a priori assumptions about the genes involved in our disease system of interest, and it has been a very successful approach for identifying novel deafness genes. In contrast, gene-driven mutagenesis starts with an identified gene and uses a variety of tools, some of which we discuss below to introduce targeted changes into the gene of interest.
In addition, researchers have utilised the many available mouse inbred strains, many of which differ in their predisposition to hearing loss [14]. It is possible to genetically map and identify loci that underlie the differences in auditory function between strains. Overall, there is a multitude of approaches that has underpinned the discovery of novel deafness loci and auditory mechanisms and underlines the utility of the humble mouse in auditory research.
Phenotype-driven screens for ARHL gene discovery
Given the relative ease by which hearing can be tested in the mouse using Click-box or Auditory-evoked Brainstem Response (ABR) tests, phenotype-driven screens of ENU-induced mouse mutants have been employed in many centres for the identification of models of hearing loss [15]. This approach has been extremely successful, leading to the identification of many deafness genes in the mouse, which have subsequently been shown to also be required for hearing in humans [13]. However, given the costs involved with housing mice these usually involve screening young animals (≤ 3 months of age) and thus, the majority of identified models had either congenital or early-onset phenotypes.
In order to uncover new genes associated with age-related auditory function we have extended this approach to include screening of aged mutant mice. The MRC Harwell Ageing Screen is the first large-scale project (~20,000 mice) to employ ENU mutagenesis and phenotyping to generate mouse models of age-related human disease. Large pedigrees of mutagenised mice are bred and enter a phenotyping pipeline comprising recurrent assessment up to 18 months of age across a wide range of disease areas, including diabetes and metabolism, neurobehaviour, bone analysis, renal function, cardiac disease, liver function, and sensorineural (vision and hearing) (Figure 1A).
The hearing screen includes recurrent Click-box and ABR phenotyping at three, six, nine and twelve months of age to enable the identification of models with ARHL. Once we have identified a pedigree with a confirmed hearing loss phenotype, we can then sequence the genomes of the affected mice to identify the underling mutation and gene (Figure 1B). This programme is beginning to identify many mutations and genes causing late-onset phenotypes, including hearing loss (http://www.mousebook.org/). In many cases we are identifying novel hearing loss genes, and we can integrate this information with studies of potential loci involved in human presbyacusis that are emerging from GWA studies in the human population.
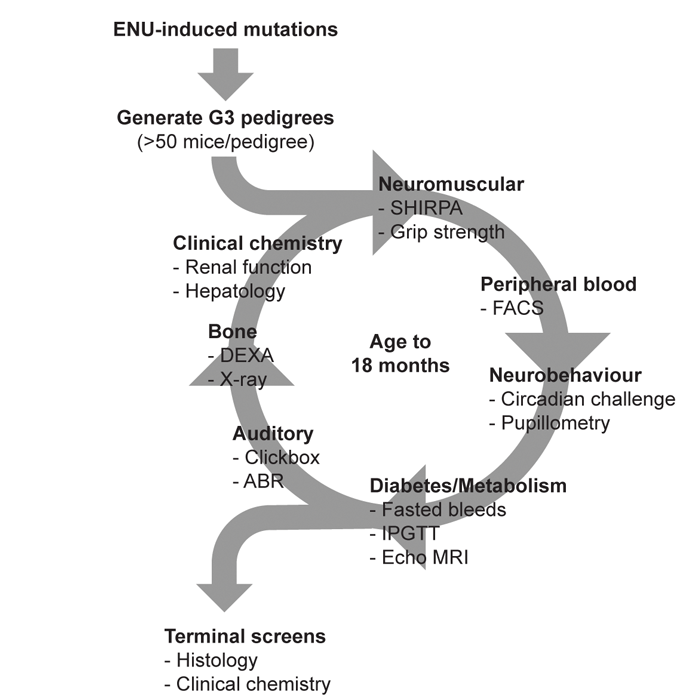
Figure 1A.
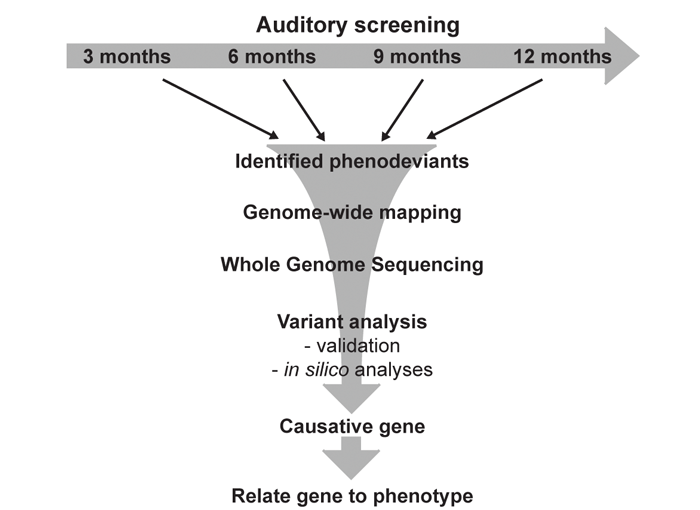
Figure 1B.
Gene-driven approaches for discovery and validation
As mentioned earlier, several genetic studies have been undertaken into adult hearing function in humans, each identifying a list of candidate genes [5-9]. Unfortunately, none of these reach genome-wide significance nor is there replication between studies. Therefore to determine which of these genes are required for auditory function a secondary validation approach is needed.
Arguably, the most effective way of exploring the requirement of these genes within the auditory system is to generate mouse knock-out models for each potential ARHL gene and test hearing function, but this is not a trivial task. However, resources are available, for example the International Mouse Phenotyping Consortium (IMPC) aims to produce a functional knock-out mouse for every known coding gene [16]. Each knock-out model produced enters a broad-based phenotyping pipeline that consists of many tests, covering a number of disciplines, carried out at defined timepoints over the first 16 weeks of life.
All the phenotyping data generated is available to view online (https://www.mousephenotype.org/). Hearing function is assessed using an ABR test at 14 weeks of age and as such will only identify relatively early-onset auditory phenotypes. However, these models are available to the scientific community allowing further characterisation at an older age. Advances in next generation sequencing methodologies now allow rapid and cost effective exome sequencing – the 1% of the genome that encodes proteins. Thus the sequencing of ARHL patients and familial cases has become a realistic possibility. However, these sequencing projects will identify a plethora of coding lesions, many of which will be non-pathogenic. The challenge will be to tease out the pathogenic mutations.
Whilst computer-based prediction programmes and in vitro assays can be used to assess the likely effects of a mutation on protein function, a model is still required to validate the lesion and to allow disease progression to be characterised. A recent development that will help facilitate the generation of such models is CRISPR/Cas9 genome-editing technology [17]. This technique has the potential to produce mouse models carrying specific gene mutations identified in human patients. In conclusion, integrated human and mouse approaches will lead to a more profound understanding of the genetics underlying diseases of ageing e.g. ARHL.
The study of mouse models, whether they are chemical-induced, knock-outs, or targeted CRISPR/Cas9 mutants, will provide essential information regarding the molecular and cellular processes occurring during ageing. This increased understanding will lay the foundation necessary for developing therapeutic strategies to prevent or ameliorate age-related functional decline.
References
1. Bainbridge KE, Wallhagen MI. Hearing loss in an aging american population: extent, impact, and management. Annu Rev Public Health 2014;35:139-52.
2. Christensen K, Frederiksen H, Hoffman HJ. Genetic and environmental influences on self-reported reduced hearing in the old and oldest old. J Am Geriatr Soc 2001;49(11):1512-7.
3. Gates GA, Couropmitree NN, Myers RH. Genetic associations in age-related hearing thresholds. Arch Otolaryngol Head Neck Surg 1999;125(6):654-9.
4. Karlsson KK, Harris JR, Svartengren M. Description and primary results from an audiometric study of male twins. Ear Hear 1997;18(2):114-20.
5. Fransen E, Bonneux S, Corneveaux JJ, et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. Eur J Hum Genet 2014;Jun 18. doi: 10.1038/ejhg.2014.56. (Epub ahead of print).
6. Friedman RA, Van Laer L, Huentelman MJ, et al. GRM7 variants confer susceptibility to age-related hearing impairment. Hum Mol Genet 2009;18(4):785-96.
7. Girotto G, Pirastu N, Sorice R, et al. Hearing function and thresholds: a genome-wide association study in European isolated populations identifies new loci and pathways. J Med Genet 2011;48(6):369-74.
8. Nolan LS, Maier H, Hermans-Borgmeyer I, et al. Estrogen-related receptor gamma and hearing function: evidence of a role in humans and mice. Neurobiol Aging 2013;34(8):2077.
9. Van Laer L, Huyghe JR, Hannula S, et al. A genome-wide association study for age-related hearing impairment in the Saami. Eur J Hum Genet 2010;18(6):685-93.
10. Schuknecht HF. Presbycusis. Laryngoscope 1955;65(6):402-19.
11. Schuknecht HF, Gacek MR. Cochlear pathology in presbycusis. Ann Otol Rhinol Laryngol 1993;102(1 Pt 2):1-16.
12. Ohlemiller KK. Age-related hearing loss: the status of Schuknecht’s typology. Curr Opin Otolaryngol Head Neck Surg 2004;12(5):439-43.
13. Brown SD, Hardisty-Hughes RE, Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat Rev Genet 2008;9(4):277-90.
14. Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics 2000;70(2):171-80.
15. Hardisty-Hughes RE, Parker A, Brown SD. A hearing and vestibular phenotyping pipeline to identify mouse mutants with hearing impairment. Nat Protoc 2010;5(1):177-90.
16. Brown SD, Moore MW. Towards an encyclopaedia of mammalian gene function: the International Mouse Phenotyping Consortium. Dis Model Mech 2012;5(3):289-92.
17. Wang H, Yang H, Shivalila CS, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013;153(4):910-8.
Declaration of Competing Interests: None declared.



