Introduction
Congenital nasal pyriform aperture stenosis (CNPAS) is a rare cause of neonatal upper airway obstruction which was first described by Douglas in 1952 [1]. The first radiological description of CNPAS was described by Ey et al in 1988 [2]. It was not until 1989 that Brown et al described the first clinical case of CNPAS [3]. Its aetiology is unknown but CPNAS is associated with a mutation in the SHH gene in some cases. Due to the association with other anomalies, its true incidence is unknown.
It can occur as an isolated anomaly or be associated with other midline defects such as solitary median maxillary central incisor syndrome (SMMCI). This syndrome can include holoprosencephaly, pituitary, cardiac and urogenital abnormalities.
As neonates are obligate nasal breathers, patients with CNPAS present to ENT services with symptoms associated with reduced nasal air flow. Clinical features include tachypnoea, poor feeding, failure to thrive, pectus excavatum, cyclical episodes of apnoea / cyanosis and resistance felt whilst passing a nasogastric tube in either nostril.
“It is essential that children with CNPAS are assessed for other associated malformations and should routinely have a cardiology review, endocrine assessment and a renal ultrasound.”
Management of CNPAS is dependent upon the individual infant and certain key clinical factors including growth, feeding and sleeping patterns and the presence or absence of cyanotic events. Initial management includes establishing the diagnosis and assessing the severity of symptoms. A craniofacial computed tomography (CT) scan is the investigation of choice (Figure 1) and a pyriform aperture width (PAW) of <11mm is diagnostic of pyriform aperture stenosis. As a rule children with a PAW of less than or equal to 7mm usually require surgery whereas those with a PAW of >7mm may be managed conservatively.
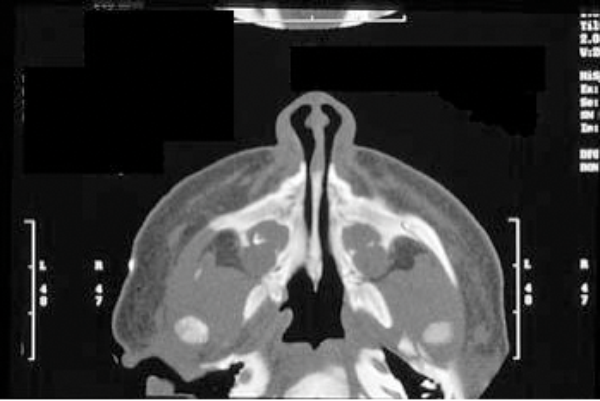
Figure 1: A craniofacial CT scan demonstrating CNPAS.
Baseline sleep studies (pulse oximetry and / or TOSCA) are also helpful in quantifying the degree of respiratory obstruction. It is essential that children with CNPAS are assessed for other associated malformations and should routinely have a cardiology review, endocrine assessment and a renal ultrasound.
Where the infant is thriving or with minimal symptoms then conservative management is recommended. Parents should be advised that symptoms may deteriorate with upper respiratory tract infections and during these a short course of nasal decongestants may prove helpful. Children with significant airway symptoms affecting the above parameters require surgical intervention. Here we describe our surgical method of managing children with CNPAS performed over 10 years at the Royal Hospital for Sick Children, Glasgow.
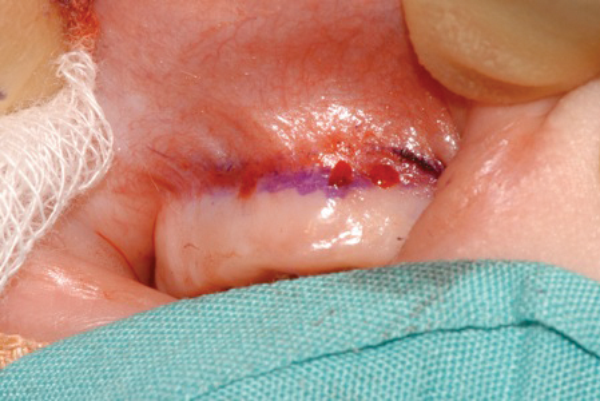
Figure 2: The stenosed pyriform aperture is approached through a sublabial incision.
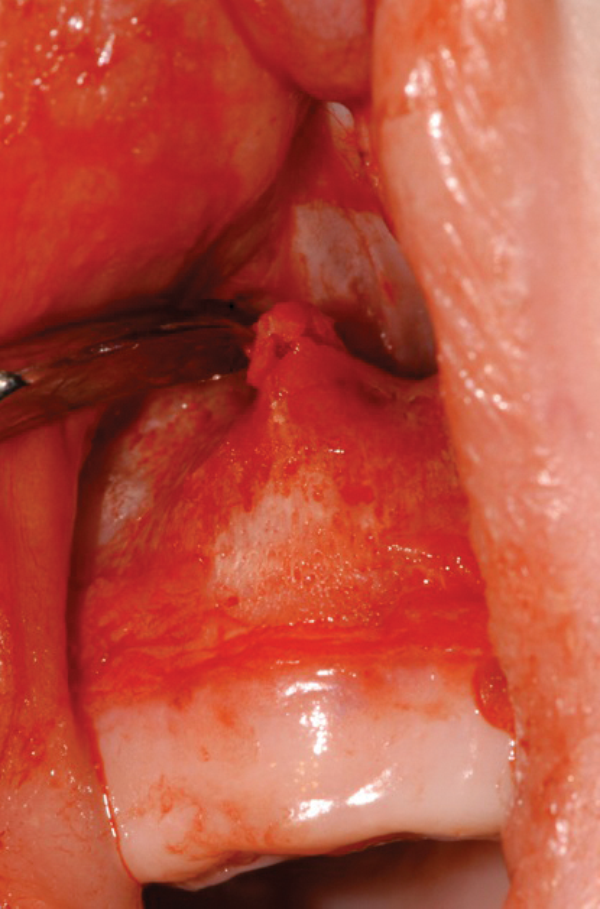
Figure 3: A mucoperiosteal flap is raised to expose the bony stenosis bilaterally.
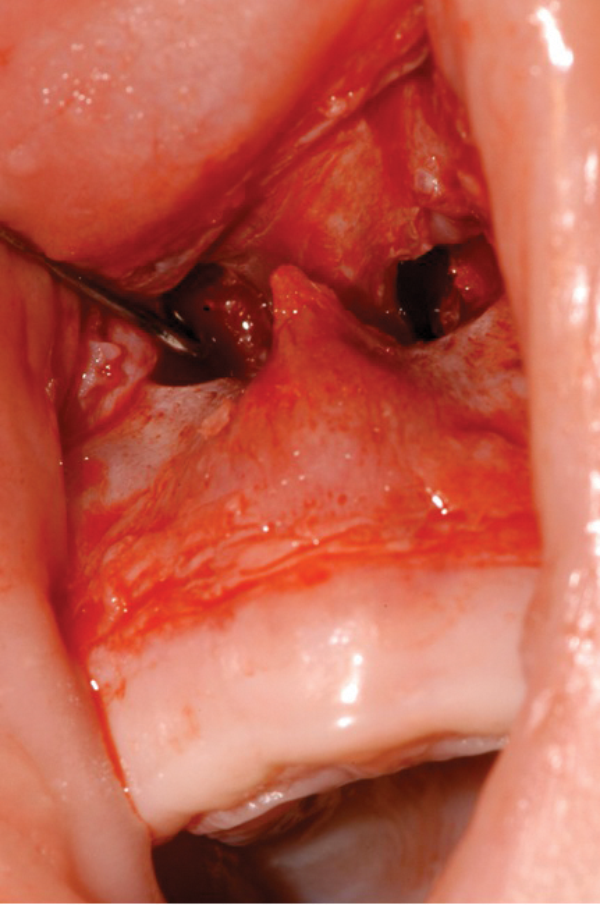
Figure 4: Excess bone is drilled away from the inferior inlet along
the floor of the nose to the lateral process of the maxilla.
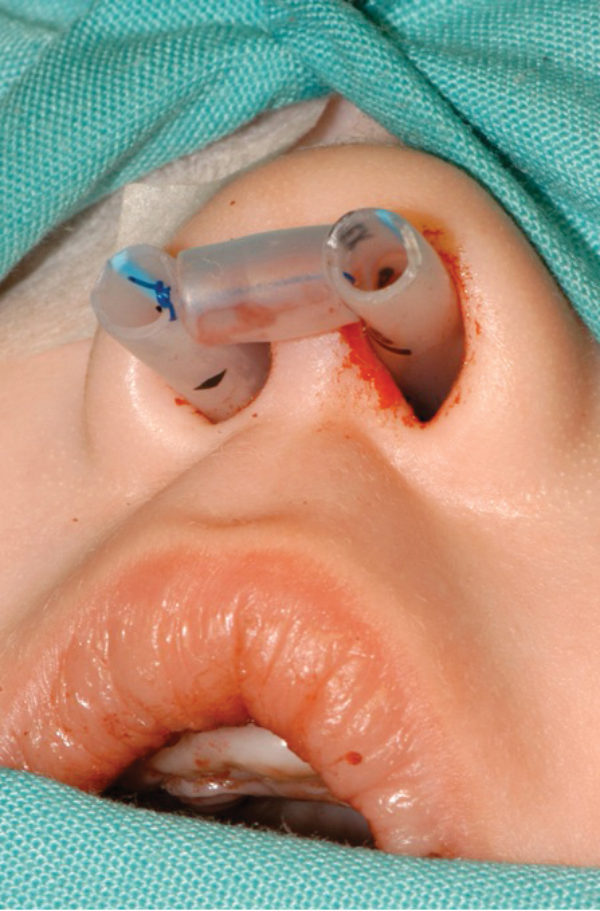
Figure 5: Nasal stents are placed to avoid restenosis.
Surgical method
- Surgery is performed under general anaesthesia.
- The stenosed pyriform aperture is approached through a sublabial incision (Figure 2).
- A mucoperiosteal flap is raised to expose the bony stenosis bilaterally (Figure 3).
- Using a 2.8mm diamond burr, excess bone is drilled away from the inferior inlet along the floor of the nose to the lateral process of the maxilla (Figure 4).
- Care is taken to avoid drilling inferiorly and posterolaterally to avoid injury to the tooth sockets/buds and nasolacrimal duct respectively.
- A guide on adequate widening of the pyriform aperture is when a 4mm rigid Hopkins rod telescope can freely pass through each pyriform aperture.
- Once resection is performed, nasal stents are placed to avoid restenosis. In our experience, a 3.5 portex endotracheal tube fashione as a nasal stent is used (Figure 5).
- Postoperative care of nasal stents included regular suctioning, nasal steroid and decongestant drops. Stents are removed 3-4 weeks later.
A total of 16 children have been treated in our unit of which 10 underwent surgical widening of the pyriform aperture. The mean age at surgery was 14 days old. Two complications following surgery were observed which included one restenosis and one patient developing granulation tissue in the nasal cavity. Apart from SMMCI other anomalies noted in our patients were choanal atresia, velocardiofacial syndrome, midfacial hypoplasia and tuberous sclerosis.
Conclusions
Congenital nasal pyriform aperture stenosis is a rare but treatable cause of neonatal upper airway obstruction. Clinical management of these children is multidisciplinary to rule out potential malformations that may coexist. Treatment is either conservative or surgical and is dependent upon the severity of symptoms.
References
1. Douglas B. The relief of vestibular nasal obstruction by partial resection of the nasal process of the superior process of the maxilla. Plast Reconstr Surg 1952;9(1):42-51.
2. Ey EH, Han BK, Towbin RB, Jaun WK. Bony inset stenosis as a cause of nasal airway obstruction. Radiology 1988;168:477-9.
3. Brown OE, Myer CM, Manning SC. Congenital nasal pyriform aperture stenosis. Laryngoscope 1989;99:86-91.
4. Blackmore K, Wynne DM. A case of solitary median maxillary central incisor syndrome with bilateral pyriform aperture stenosis and choanal atresia. Int J Pediatr Otorhi 2010;74:967-9.
5. Visvanathan V, Wynne DM. Congenital nasal pyriform aperture stenosis. A report of 10 cases and literature review. Int J Pediatr Otorhi 2012;76(1):28-30.
Declaration of competing interests: None declared.





