Branchio Oto Renal Syndrome (BOR) is an autosomal dominant disorder with branchial, otologic and renal manifestations. The presence of clinical signs varies among and within affected families. Three causative genes for BOR syndrome have been reported thus far: EYA1, SIX1 and SIX5, but the causative genes for approximately half of all BOR patients remain unknown. To make a diagnosis, the knowledge of clinical features of genetic syndromes is of paramount importance. We are presenting three of our patients with this rare syndrome: a girl who has fulfilled diagnostic criteria for BOR syndrome and two mothers with daughters. Differential diagnosis between similar syndromes with phenotypic variation is delicate, especially without genetic examinations.
Introduction
BOR syndrome (or Branchio Oto Renal Dysplasia, Melnick-Fraser Syndrome) is an autosomal dominant disorder with branchial, otologic and renal manifestations. The literature indicates a prevalence of BOR syndrome of 1:40,000 making this disorder responsible for about two percent of profound hearing impairment in children [1, 2].
Etiology: genetic background
Three causative genes for BOR syndrome have been reported thus far: EYA1, SIX1 and SIX5, but the causative genes for approximately half of all BOR patients remain unknown [3, 4]. The majority of cases of BOR syndrome are caused by mutations in EYA1 on chromosome 8q, which has 16 exons [4, 5].
Clinical features
The disease is represented by various phenotypic expressions. The presence of clinical signs varies among and within affected families. The clinical features of BOR syndrome include otological malformations with associated hearing loss, branchial cleft sinuses, cervical fistulas and renal anomalies, with range from mild hypoplasia to bilateral renal agenesis [1-4].
Otological manifestations of BOR syndrome are malformations of the outer, middle or inner ear. Hearing impairment can vary from mild to profound degree. According to the site of the lesion conductive, sensorineural and mixed hearing impairment are distinguished. In the external auditory canal stenosis of varying degree can occur, even atresia. Middle ear may be affected by various anomalies of malformation, malposition or fixation of the ossicles, and irregular size and shape of tympanic cavity. Cochlear hypoplasia, cochlear and vestibular large aqueducts or hypoplasia of the lateral semicircular channel can be found in the inner ear [1-3].
There are several types of malformations of the branchial arches in BOR including cupping of the outer ear, ear pits and tags of skin in front of the ear, and cysts or fistulas on the neck [2, 3]. Renal anomalies range from mild hypoplasia to bilateral renal agenesis which is incompatible with living [1, 5].
Diagnosis
To make a diagnosis, the knowledge of clinical features of genetic syndromes is of paramount importance. Complete evaluation includes paediatric examination, especially nephrological, bone and soft tissue radiological visualisation, i.e. computed tomography and nuclear magnetic resonance and finally genetic tests in cases where a hereditary disorder is suspected or identified [1-6].
The wide spectrum of phenotypic findings associated with EYA1 mutations can make the diagnosis of BOR syndrome difficult. An additional problem is genetic heterogeneity; mutations in other genes cause a similar phenotype [5, 6, 8].
“The wide spectrum of phenotypic findings associated with EYA1 mutations can make the diagnosis of BOR syndrome difficult.”
Interesting research was recently published by Krug and colleagues. Their report refers to a screening of these three genes in a cohort of 140 patients from 124 families with BOR. Krug and coauthors identified 36 EYA1 mutations in 42 unrelated patients, two mutations and one change of unknown significance in SIX1 in three unrelated patients, but no mutation in SIX5. They did not find correlation between genotype and phenotype, and observed a high phenotypic variability between and within BOR families. They also show the difficulty in establishing a molecular diagnosis strategy in BOR syndrome: the screening focusing on patients with typical BOR would detect a mutation rate of 76%, but would also miss mutations in 9% of patients with atypical BOR [8].
Renal ultrasonography or intravenous pyelography is essential to determine the extent of renal involvement with functional investigation. Diagnosis and treatment is the responsibility of paediatricians and nephrologists [1].
Differential diagnostic considerations
There are a few syndromes very similar to BOR, these include Branchio Otic syndrome (BOS), Branchio Oto Ureteral syndome (BOU), Branchio Oculo Facial syndrome (BOFS) and Oto Facio Cervical syndrome (OFC). Differential diagnosis between these similar syndromes with phenotypic variation is delicate especially without genetic examinations [9].
Case outline
We are presenting three of our patients with this rare syndrome. First, we present a girl, aged four years at diagnosis, who has fulfilled BOR diagnostic criteria manifesting three cardinal features: unilateral renal hypoplasia, bilateral cervical branchial cysts and a hearing loss. Family history was negative for hearing loss, neck fistula and kidney problems. She has a younger healthy brother. She has achieved regular milestones, except development of speech, which was delayed and non-intelligible for the unknown listeners. Her intelligence and growth were normal at the age of four years. In-utero presence of unilateral renal hypoplasia was noticed. Lateral fistula on the left neck-side was operated on at the age of three years. Preauricular pits were present on both sides. Otoscopic and rhinoscopic findings were regular bilaterally. Throat was showing velopharyngeal insufficiency without having signs of submucous cleft palate as a minor criteria. Tympanogram was type A on both sides and acoustic reflex was absent bilaterally at all probe intensities. Transient otoacoustic emissions were not registered. Tonal audiometry showed a conductive hearing loss bilaterally with pure tone average about 75 dB for air conduction. Brainstem evoked auditory potentials confirmed this finding. Caloric test showed regular function of the both labyrinths. Computerised tomography of temporal bones confirmed malformations in both middle ears (Figure 1). Genetic investigations in the UK have confirmed the presence of BOR syndrome registering the EYA 1 mutation at 8q chromosome (Figure 2).

Figure 1: Audiology findings belonging to a four year old girl with Branchio Oto Renal syndrome
who has conductive hearing loss (some images scanned from originals).
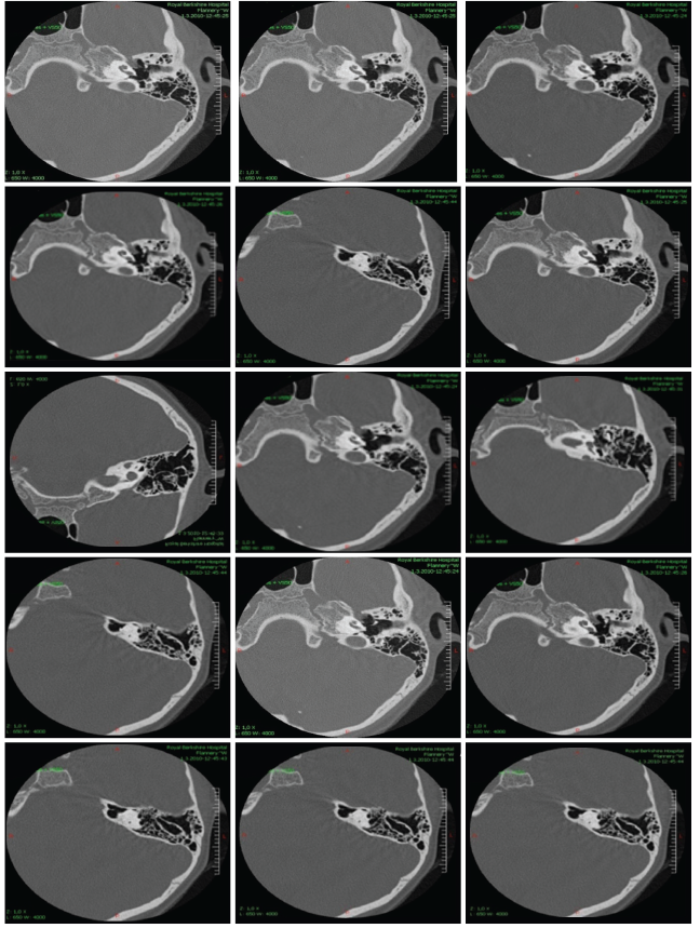
Figure 2: Computerised tomography of temporal bones belonging to a four year old girl with Branchio Oto Renal
syndrome who has conductive hearing loss as a conseqence of ossicle malformations.
The girl has started with auditory training using hearing aids for air conduction. Speech is now excellent and intelligible; she is a bilingual speaker and an excellent pupil in regular school [9].
The other cases concern two families. The first, a mother and daughter with sensorineural hearing loss, lateral neck fistula and renal hypoplasia. The mother has a mild severity sensorineural hearing loss, but the daughter has a profound sensorineural hearing loss and bilateral low set small, malformed ears. She has undergone cochlear implantation (Figure 3). Another family with sensorineural hearing loss comprises a mother with profound hearing loss, lateral neck fistula and severe kidney failure who is waiting for kidney transplantation. Her daughter has mild sensorineural hearing loss and mild unilateral kidney hypoplasia without branchial anomalies (Figure 4). Diagnosis of BOR syndrome in these two families was performed on the basis of clinical findings, without genetic confirmation.
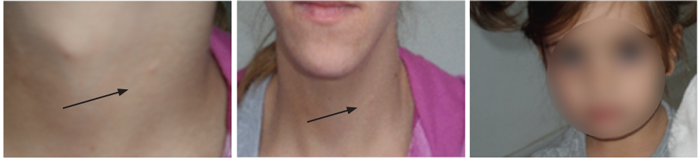
Figure 3: Mother with profound sensorineural hearing loss, lateral neck remnants, kidney
hypoplasia and daughter with the same signs except branchial remnants.

Figure 4: Mother with middle degree sensorineural hearing loss, lateral neck remnants, kidney hypoplasia and
daughter with profound sensorineural hearing loss, malformed auriculas, branchial remnants and kidney hypoplasia.
Conclusion
Discovering diagnosis of disorders in the context of genetic syndromes and the cause is important for rehabilitation planning, prognostic assessment and family planning. Rare cases of hereditary syndromes with hearing problems provide more knowledge about the structure of hereditary hypoacusis forms in the population. The syndromal forms reflect a complex genetic basis of the processes of sound perception.
References
1. Chun-Hui Tsai A, Vallee SE. In: Bluestone CD, (Ed.). Paediatric Otolaryngology, 4th edn. Phialdelphia, USA; Elsevier Science; 2003:37-59.
2. Acierno SP, Waldhausen JH. Congenital cervical cysts, sinuses and fistulae. Otolaryngol Clin North Am 2007;40(1):161-76.
3. Orten DJ, Fischer SM, Sorensen JL. Branchio-oto-renal syndrome (BOR): novel mutations in the EYA1 gene, and a review of the mutational genetics of BOR. Hum Mutat 2008;29(4):537-44.
4. Morisada N, Nozu K, Iijima K. Branchio-oto-renal syndrome: comprehensive review based on nationwide surveillance in Japan. Pediatr Int 2014;56(3):309-14.
5. Okada M, Fujimaru R, Morimoto N, Satomura K, Kaku Y, Tsuzuki K, Nozu K, Okuyama T, Iijima K. EYA1 and SIX1 gene mutations in Japanese patients with branchio-oto-renal (BOR) syndrome and related conditions. Pediatr Nephrol 2006;21(4):475-81.
6. Sanggaard KM, Rendtorff ND, Kjaer KW, et al. Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses. Eur J Hum Genet 2007;15(11):1121-31.
7. Krug P, Morinière V, Marlin S, et al. Mutation screening of the EYA1, SIX1, and SIX5 genes in a large cohort of patients harboring branchio-oto-renal syndrome calls into question the pathogenic role of SIX5 mutations. Hum Mutat 2011;32(2):183-90.
8. Matsunaga T, Okada M, Usami S, Okuyama T. Phenotypic consequences in a Japanese family having branchio-oto-renal syndrome with a novel frameshift mutation in the gene EYA1. Acta Otolaryngol 2007;127(1):98-104.
9. Ječmenica J, Bajec-Opančina A. Branchiootorenal and branchiooculofacial syndrome. J Craniofac Surg 2015;26(1):e30-1.
Declaration of Competing Interests: None declared.


