Introduction
Cancer cases continue to increase worldwide, and head and neck cancer is a major global health issue, with an estimated global burden of over 630,000 new cases and over 350,000 deaths per year [1]. The term ‘head and neck cancer’ represents a heterogeneous group of tumours that occupy different sites within the upper aero-digestive tract, and arise from different tissues of origin, with HNSCC by far the most common. Treatment regimens depend on site of origin, histological tumour type and stage and grade of disease.
Tobacco smoking, alcohol, EBV and HPV infection are known major risk factors for different subtypes of head and neck cancer but genetics also plays a major role, as highlighted by tumour syndromes that run in families. Despite continued advances in surgical techniques, chemotherapy and radiotherapy, the overall survival rates for many subtypes of HNSCC have not improved significantly over the last 40 years and advanced / recurrent disease is still associated with a poor prognosis, as is true for multiple of other types of cancer. In recent years research has shifted to focus on cancer genetics, aiming to discover novel treatments to improve outcomes.
Signalling pathways in health
Cells in most tissues are in a constant state of turnover, renewal and repair, largely driven by responses to internal and external cues. These signals are secreted and received by the same cell (intracrine / autocrine), local acting (juxtacrine / paracrine) or excreted, acting on distant target cells (endocrine). Chemical cues (hormones, growth factors, neurotransmitters, cytokines) bind to cell surface receptors, activating them and triggering an intracellular signalling pathway resulting in a response at the molecular genetic level – typically changes in protein transcription and translation. These pathways are active in normal cellular homeostasis, but are usually tightly controlled by feedback loops. Dysregulation of cell signalling pathways is now a well-established feature of tumourigenesis, and research is currently focused on finding ways to target this issue.
Signalling pathways in disease
There are three types of proteins that regulate the cell cycle, and these correspond to the three main types of cancer susceptibility genes. Although these genes work in different ways, impaired function in any of them all result in the same thing – unregulated cell proliferation and increased mutation rate (Figure 1).
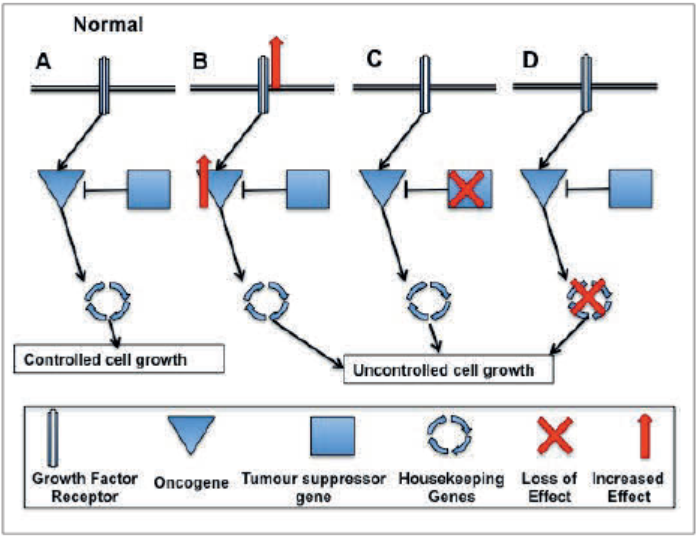
Figure 1: Types of genes involved in cell signalling and cancer.
A: Under normal conditions there is a balance between proteins that promote cell growth and proliferation when stimulated by growth factors or high energy levels (proto-oncogenes), and those that prevent it (suppressors). Multiple mechanisms are in place to ensure that the receptors and proto-oncogenic signalling pathways are switched off again once the cell has responded to the initiating signal. Downstream from this point there are a series of ‘checking mechanisms’ designed to ensure that any mistakes made during replication of genetic material are either corrected, or if too damaged, the cell apoptoses and dies.
B: Oncogenes. The proteins function in the same way, but their effect is potentiated, frequently by gene amplifications, chromosomal translocations or ‘gain of function’ dominant mutations which increase activity of the protein, resulting in constitutional activation of downstream signalling pathways.
C: Tumour suppressor genes. In tumourigenesis, their suppressive function is lost, (through inactivating ‘loss of function’ mutations or chromosomal deletions) which allow the cell cycle to proceed unchecked.
D: Stability or caretaker genes are a diverse group, including repair genes (required to ‘proof read’ DNA during replication, removing any mistakes) and genes involved in mitotic recombination and chromosomal segregation during mitosis. Under normal circumstances badly damaged cells undergo apoptosis. In cancer this mechanisms is impaired, allowing the badly mutated cells to persist and replicate.
These mutations may be germline, resulting in hereditary predisposition to disease and manifesting as familial disease; or somatic, arising within a single tissue of the body and causing sporadic disease. Examples of germline mutations include FMTC and MEN (which exhibit autosomal dominant inheritance) and paraganglioma-pheochromocytoma syndromes (autosomal dominant inheritance complicated by paternal imprinting and variable penetrance) [2].
The underlying mutations for these and many other tumours were discovered using genome-screening techniques, comparing results between affected and unaffected relatives. Detection of somatic mutations has been revolutionised by the development of high throughput NGS techniques, which when recently applied to HCSCC demonstrated frequent somatic mutations in signalling pathway genes, with very high rates of mutation seen in smokers and heavy drinkers [3].
These studies also revealed far lower mutation rates in HPV positive patients (who have a better prognosis compared to HPV negative patients) [4] demonstrating that the local environment has an impact on mutation rates and therefore risk of tumour formation. These studies have also demonstrated how heterogeneous tumour tissue can be – i.e. not every cell will express the same mutation simultaneously – an important fact to consider when designing targeted therapies, since inhibiting the effect of one mutation may simply select for clonal expansion of a second subset of tumour cells which do not express that mutation, and hence treatment will fail [5].
Mitogenic signalling pathways
One of the best studied is probably the MAPK and PI3K/AKT/mTOR pathways – which are triggered by binding of growth hormones at the cell surface (Figure 2). In normal tissues, these pathways regulate cellular homeostasis, ensuring that cells only perform anabolic tasks (cell cycle progression) when energy supplies are appropriate, while switching to catabolism in nutrient deplete circumstances. Binding of growth hormones or neurotransmitters triggers cascades of signalling pathways within the cell, affecting transcription of factors that may promote cell survival and angiogenesis.
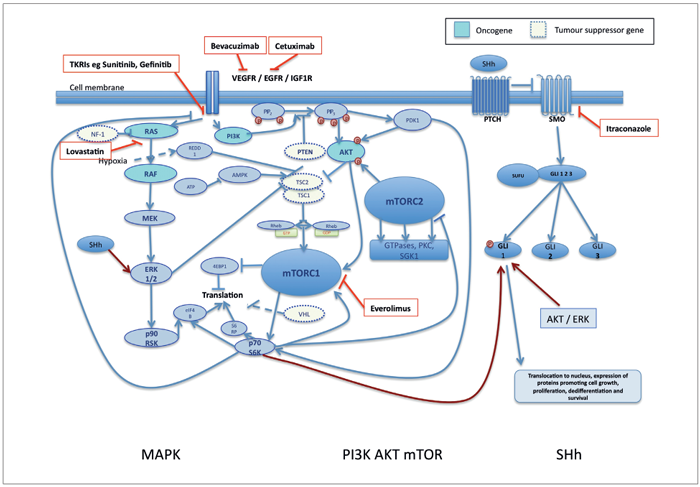
Figure 2: Cell signalling pathways implicated in tumourigenesis. Examples of signalling pathways common to many cancers. Non-canonical cross activation is shown in dark red. The mTOR and MAPK pathways are triggered by binding of growth factors to cell surface receptors, but can also be activated by cross talk (via other pathways e.g. SHh) further down. The SHh pathway is activated by binding of SHh ligand to the cell surface PTCH receptor, which then releases its negative control over SMO, enabling it to trigger the downstream pathway. This pathway is commonly activated by cross talk from other pathways, as shown. Licensed inhibitors for specific targets are shown in red. There are multiple feedback loops not shown for simplicity. These can cause major problems with treatment - for example, P70S6K, once activated, negatively regulated the cell surface growth receptor, triggering internalisation and preventing further signalling. When this loop was blocked using mTOR inhibitors, the MAPK pathway was able to continue unchecked, leading to increased levels of ERK, and hence poor responses to treatment [10]. Via similar mechanisms, mTORC1 inhibitors prevent feedback inhibition of AKT. Both AKT and ERK can trigger non-canonical activation of other pathways such as SHh, again providing a mechanism of overcoming treatment.
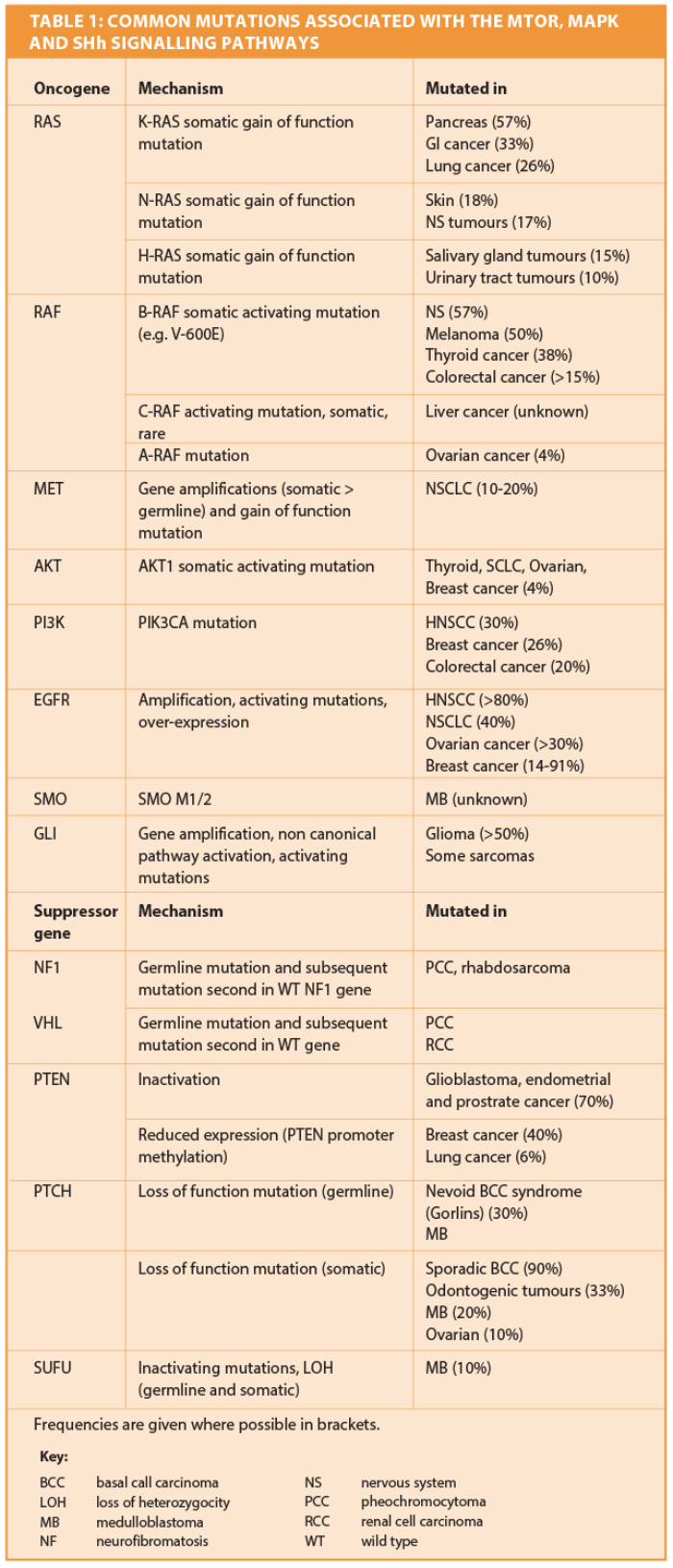
Dysfunction of aspects of the mTOR pathway have been demonstrated for many different tumours including breast, liver, ovarian, melanoma, renal cell carcinoma, haematological malignancies, and neuroendocrine tumours including MEN. Oncogenes PI3K, AKT and Rheb are commonly either mutated or highly expressed in a number of tumours, with resultant downstream pathway activation [6]. PTEN, TSC1 and TCS2 function as tumour suppressor genes, and loss of function mutations in these genes is a frequent observation in many tumours, including HNSCC [4]. Mutations are also common in the MAP kinase signalling pathway, where a number of enzymes have been shown to function as oncogenes (Table 1). Overexpression of EGFR is seen in many tumours, including HNSCC, where over 80% demonstrate abnormal levels [7] and the same is true for VEGFR.
Overexpression is not always a result of activating mutations, but can also be triggered by hypoxia and non-canonical activation of signalling pathways [7]. The rapid growth of tumours necessitates an increased blood supply compared to normal tissues. Angiogenesis is normally a tightly regulated process, but the up-regulation of signalling pathways, combined with hypoxia often seen in larger tumours creates an environment that promotes an ‘angiogenic switch’ towards new blood vessel formation – a process in which VEGFR and VEGF are key players [8, 9].
Other signalling pathways
While this pathway is particularly well described in cancer, other pathways (including but not limited to) angiogenesis (VEGFR) [11], stem cell / embryological pathways (SHh, NOTCH and Wnt) [5], hypoxia signalling pathways (HIF) [12] and heat shock protein (HSP) [13] are also frequently dysregulated. These pathways can be affected by mechanisms described above, plus non-canonical activation by ‘cross talk’ from other pathways [14].
In some tumours there is evidence that different signalling pathways may be important during different stages of disease progression, which has implications for treatment regimens [15]. Another important consideration is that most tumours contain populations of cancer stem cells, which are more resistant to chemotherapy and radiotherapy, which act as a form of selection for highly resistant stem cells which can then undergo clonal expansion, triggering relapse. There is mounting evidence to suggest that they contribute to drug resistance, EMT, recurrence and metastasis [5].
Current treatments
In recent years a number of strategies have been tried to effectively target these pathways in cancer cells. A brief summary of treatments currently licensed is given below. The aim of all of these treatments is to inhibit effects of over-expressed oncogenes, restore function of under-expressed TSGs, promote apoptosis of tumour cells and impair factors that promote cell division and EMT, while minimising impact on off target tissues.
Monoclonal antibodies
These typically target cell surface receptors trying to prevent activation of downstream signalling pathways. Licensed examples include antibodies to EGFR (Cetuximab for HNSCC and colorectal cancer, Herceptin licensed for breast cancer) or VEGFR (Bevacizumab for RCC, metastatic colorectal cancer, glioblastoma). They are given by IV infusion and are more specific in action than other targeted agents, blocking either the receptor or its ligand. Cetuximab has been shown to improve overall survival for HNSCC when given with radiotherapy, and is recommended in patients in whom platinum based chemotherapy is contraindicated.
Receptor tyrosine kinase inhibitors
These orally available small molecules are designed to inhibit the intracellular kinase domain of the receptors. This group includes inhibitors of EGFR (Gefitinib and Erlotinib licensed for NSCLC) and VEGFR (Sunitinib – a multi-tyrosine kinase that targets several growth factor receptors and is licensed for metastatic RCC, pancreatic NETS and advanced GIST) plus Vandetinib (which inhibits EGFR, VEGFR and RET and is used to treat metastatic medullary thyroid cancer). Response to treatment is often mutation specific – for example in NSCLC patients carrying common somatic EGFR mutations RTKIs are superior to chemotherapy as first line treatment, but some rare mutations confer resistance.
Small molecule serine threonine kinase inhibitors
These orally available drugs target key protein(s) within the intracellular signalling pathway downstream of the initiating cell surface receptor. Examples in clinical practise include the mTOR inhibitors Everolimus against advanced breast and pancreatic cancer, Temsirolimus against RCC and the B-RAF inhibitor Vemurafenib against metastatic melanoma.
Receptor mimics
Ziv-aflibercept (Zaltrap) binds circulating VEGF preventing it binding and activating VEGFR. This is given IV and is currently used for advanced colorectal cancer non responsive to conventional treatment. There are many alternative therapies still at trial / experimental stages. These include recombinant antibody toxins, antisense oligonucleotides and ribozyme mediated suppression of gene expression, oncolytic viruses targeting tumour cells or angiogenesis and gene therapy, delivered by a range of vectors.
Cancer genetics in rare tumours
With rare tumours that do not appear to have a hereditary component, it is not as easy to unpick the underlying genetic defects promoting tumourigenesis. For example, olfactory neuroblastoma, a rare neuroendocrine tumour thought to arise from the olfactory neuroepithelium at the top of the nasal cavity has no known risk factor or genetic predisposition. Initially thought to be similar to neuroblastoma, it is now known to have more in common with other tumours from the small round blue cell group (including Ewing’s sarcoma and NK/T cell lymphoma) [16]. ONB typically presents with non-specific rhinological symptoms and therefore disease may be advanced before the diagnosis is made. Numerous studies have shown that low stage and grade disease responds well to current gold standard treatment of craniofacial resection / endoscopic resection plus post op RT / Chemo RT [17]. However, this tumour has a tendency to recur sometimes many years after the initial diagnosis, and patients with advanced or metastatic disease at the time of diagnosis still have a poor prognosis.
In keeping with findings on other tumours, IHC studies confirmed high levels of the tyrosine kinase receptors VEGFR, IGF1R, and neurotrophin receptors in ONB compared to normal nasal mucosa. High levels of PDGFR is demonstrable in endothelial and other surrounding cells in metastatic disease [18]. These TKRs receptors all activate the mTOR and MAPK signalling pathways. Subsequent studies revealed high levels of proteins linked to hypoxia (HIF1, anti-apoptosis (BCL-2) oncogenic transformation (c-MYC) and developmental pathways (SHh). These proteins are known to cross react – for example, HIF1, once activated increases transcription of BCL-2 and VEGFR, which leads to further increase in HIF1 expression in a feedback loop involving the mTOR pathways. Both the MAP kinase cascade and the SHh pathway cause increased c-MYC expression, which triggers similar feedback loops to both pathways, ultimately promoting angiogenesis (VEGFR) and inhibiting apoptosis (BCL2).
Cytogenetic studies and whole genome sequencing have provided more evidence to suggest the MAPK and mTOR pathways are involved in tumourignesis in ONB [19]. Further evidence comes from two recent cases of advanced metastatic ONB which demonstrated prolonged response to palliative treatment with the VEGFR inhibitor sunitinib [18] and the anti VEGFR monoclonal Bevacizumab (Avastin) [20] both of which were well tolerated with no significant side-effects, and were able to halt disease progression for some months before the tumours progressed again.
Research is currently focused on novel agents with the potential to target the mTOR and SHh pathway simultaneously in this disease. These treatments will facilitate understanding of the feedback loops and cross talk between the two pathways, which may guide future therapy development. The findings will be applicable to other tumours that show a similar pattern of pathway activation, and may ultimately provide better treatment for advanced disease. Nasal disease is more amenable to topical treatments than cancers at almost any other site (other than skin). It may in future be possible to apply topical treatment at the end of surgical resection, targeting residual cells, blocking CSC activity and triggering apoptotic pathways in tumour cells thereby negating the risk of long-term recurrence currently associated with this disease. Similar approaches are being used for other rare diseases – for example dual targeting of mTOR and HSP90 in metastatic pheochromocytoma / paraganglioma syndromes [15].
Summary
Targeted inhibition of signalling pathways shows great promise for cancer therapy. Studies combining both monoclonal antibodies or TKIs with radiotherapy for advanced head and neck cancer have shown strong evidence of synergism, and there are now multiple studies targeting more than one pathway simultaneously, with the aim of eradicating CSCs as well as the main bulk of tumour tissue [14]. It is likely in the future that targeted therapies will become highly personalised to specifically target an individual patient’s underlying germline and somatic mutations, avoiding inherent resistance associated with certain mutations by ensuring only appropriate treatments are selected. If this does become possible, these treatment regimens may have a wider role to play in cancer therapy, and may ultimately become a first line treatment for some disorders.
Abbreviations
4EBP1 A translation initiation factor
AKT Protein kinase B
BRCA1 Breast cancer 1
EBV Epstein Barr virus
EGFR Epidermal growth factor receptor
eIF4B A transcription initiation factor
ERK Extracellular regulated MAP kinase
EMT Epithelial to mesenchymal transition
FMTC Familial medullary thyroid cancer
IGF1 Insulin growth factor receptor 1
IHC Immuno histochemistry
HIF Hypoxia inducible factor
HNSCC Head and neck squamous cell carcinoma
HPV Human papilloma virus
MAP Mitogen activated protein
MAPK Mitogen activated protein kinase
MEK Mitogen activated protein kinase kinase
MEN Multiple endocrine neoplasia mTOR Mammalian target of rapamicin
NGS Next generation sequencing
PDGFR Platelet derived growth factor receptor
PDK1 Phosphatidylinositol dependent kinase 1
PI3K Phosphatidylinositol 3-kinase
PIP Phosphatidylinositol bi and tri-phosphate
P70S6K P70 ribosomal S6 kinase (S6K1)
P90RSK P90 ribosomal S6 kinase
RAS Rat sarcoma virus oncogene
RAF Rapidly accelerated fibrosarcoma Rheb
RAS homologue enriched in brain SHh Sonic hedgehog
S6RP S6 ribosomal protein
TSC1/2 Tuberous sclerosis complex
TKI Tyrosine kinase inhibitor
TKRI Tyrosine kinase receptor inhibitor
TSG Tumour suppressor gene
TSC Tuberous sclerosis complex
Wnt Wingless / integrated
References
1. Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008. Int J Cancer 2010;127:2893-917.
2. Galan SR, Kann PH. Genetics and molecular pathogenesis of pheochromocytoma and paraganglioma. Clin Endocrinol 2013;78:165-75.
3. Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Ann Oncol 2014;25(10):1889-900.
4. Loyo M, Li RJ, Bettegowda C, et al. Lessons learned from nextageneration sequencing in head and neck cancer. Head Neck 2013;35:454-63.
5. Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: in the line of fire. Cancer Treat Rev 2012;38:589-98.
6. Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol 2010;20:427-34.
7. Yewale C, Baradia D, Vhora I, et al Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 2013;34:8690-707.
8. Roskoski R Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol 2007;62:179-213.
9. Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 2007;19:2003-12.
10. Nölting S, Garcia E, Alusi G, et al. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. J Mol Endocrinol 2012;49:79-96.
11. Goel HL, Mercurio AM. VEGF targets the tumour cell. Nat Rev Cancer 2013;13:871-82.
12. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer Metab 2014;2:3.
13. Jego G, Hazoume A Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett 2013;332:275-85.
14. Brechbiel J, Miller-Moslin K, Adjei AA. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat Rev 2014;40:750-9.
15. Giubellino A, Sourbier C, Lee MJ, et al. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PloS One 2013;8:e56083.
16. Bridge JA, Bowen JM, Smith RB. The small round blue cell tumors of the sinonasal area. Head Neck Pathol 2010;4:84-93.
17. Rimmer J, Lund VJ, Beale T, et al. Olfactory neuroblastoma: a 35-year experience and suggested follow-up protocol. Laryngoscope 2014;124:1542-9.
18. Preusser M, Hutterer M, Sohm M, et al. Disease stabilization of progressive olfactory neuroblastoma (esthesioneuroblastoma) under treatment with sunitinib mesylate. J Neurooncol 2010;97:305-8.
19. Weiss GJ, Liang WS, Izatt T, et al. Paired tumor and normal whole genome sequencing of metastatic olfactory neuroblastoma. PloS One 2012;7:e37029.
20. Dunbar EM, Pumphrey PK, Bidari S. Unexpectedly durable palliation of metastatic olfactory neuroblastoma using anti-angiogenic therapy with Bevacizumab. Rare Tumors 2012;4:e33.
Declaration of competing interests: None declared.



