Gene therapies for hearing loss are rapidly advancing and will be transitioning to clinical practice. Here, the authors explain why clinicians involved in managing these disorders need to be aware of these advances.
Genomic testing in England was significantly reconfigured in 2021 through the creation of seven genomic laboratory hubs (GLHs) to form the national Genomic Medicine Service (GMS). The GMS is underpinned by the NHS England National Genomic Test Directory [1]; this specifies which GLHs are commissioned to provide each testing indication, the testing methodology, and the eligibility criteria for each indication. The aim of the GMS is to provide consistent and equitable access to genomic testing across the whole of England.
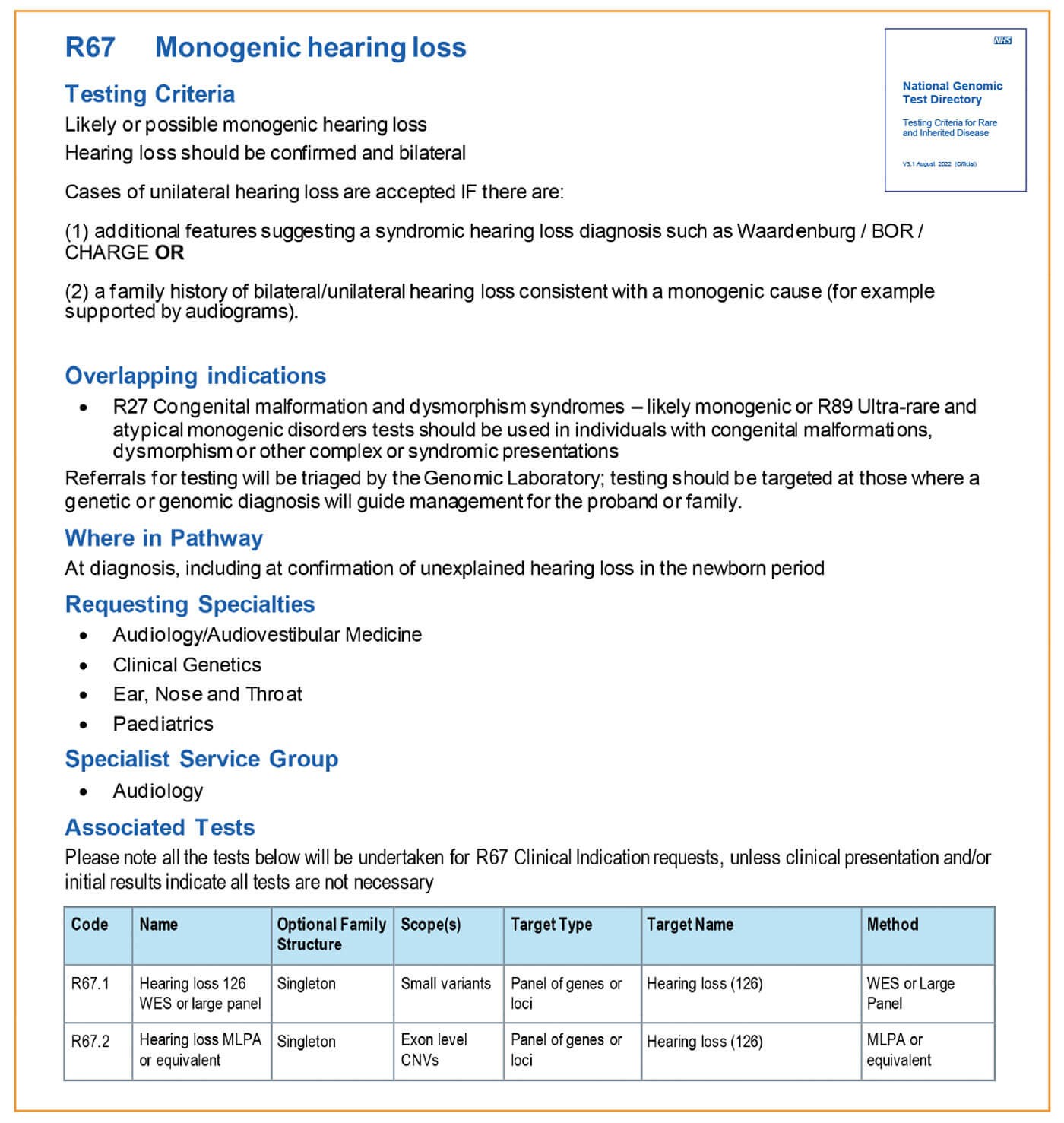
In the past, genomic testing for deafness in England was extremely limited: for most patients, single gene testing for connexin 26/30 (GJB2/GJB6 genes) was the only readily available test, meaning many families remained undiagnosed. Genomic testing for deafness is now delivered through a single specialist testing indication, R67 monogenic hearing loss, delivered by the North West and North Thames GLHs. R67 currently includes testing for variants in 115 genes, including GJB2 and GJB6, and can be requested by audiology/audiovestibular medicine, ear, nose and throat, paediatrics and clinical genetics.
Targeted testing for the m.1555A>G variant associated with aminoglycoside-induced hearing loss is available through a separate testing indication, R65 (aminoglycoside exposure posing risk to hearing). This is a core test, delivered by all seven GLHs.
When should an R67 test be considered?
R67 testing is primarily indicated for people with confirmed bilateral sensorineural deafness. Unilateral deafness is less likely to have a genetic basis, unless the patient has additional syndromic features (for example, pigmentary features of Waardenburg syndrome or branchial abnormalities for branchiootorenal syndrome), or there is a family history consistent with a genetic cause.
What is involved in the testing?
R67 testing is delivered by next generation sequencing (NGS), allowing multiple patients to be tested simultaneously for variants in many genes. The gene content of GMS panels is defined within Genomics England’s PanelApp (panelapp.genomicsengland.co.uk), an expertly curated publicly available resource. Current R67 genes include those associated with non-syndromic and some syndromic conditions (particularly where deafness may be the presenting feature). NGS permits detection of most types of genetic variation, from the substitution of single bases in the DNA sequence, to changes in copy number; that is, deletions or duplications of exons or even entire genes.
SEE ALSO

Every person’s genome contains hundreds of differences compared to reference sequences. Most of these are common benign variants with no link to clinical presentations. These may be easy to assess, but rare variants constitute a significant proportion of all variants and often require further investigation. The Association for Clinical Genomic Science’s best practice guidelines [2] are used to classify variants on a range of criteria, including presence in the literature, co-segregation with disease within families, functional studies, and computational predictions of pathogenicity. Variants are assigned to classes 1-5, representing benign, likely benign, uncertain significance, likely pathogenic, and pathogenic respectively.
"R67 currently includes testing for variants in 115 genes, including GJB2 and GJB6, and can be requested by audiology/audiovestibular medicine, ear, nose and throat, paediatrics and clinical genetics"
What variants are reported?
Pathogenic or likely pathogenic variants have a high probability of being the cause of the patient’s deafness, where they occur in keeping with the inheritance pattern known for the gene or specific variant (one altered copy for dominant or X-linked recessive; two altered copies for autosomal recessive). Certain variants of uncertain significance (VUS) may also be reported, particularly where further investigations, such as testing family members, may provide additional evidence to upgrade the classification to likely pathogenic. Benign and likely benign variants are not included on reports, and neither are most VUS, due to lacking evidence for pathogenicity.
What are the implications of a molecular genetic diagnosis for deafness?
If a potentially causative variant has been identified, further testing may be available for the patient’s family, either to assist with interpretation of the variant, or to clarify the status of individuals who may be at an elevated chance of having deafness themselves, or of having children with deafness. When a potentially causative variant has been identified, genetic counselling and/or cascade testing for the patient’s family members may be indicated, and can be arranged via referral to genomic medicine/clinical genetic services.

Role of genetic hearing and balance disorders clinics and coordinated pathways for testing and management
Ordering hearing loss gene panel tests has been extended to all parts of the country, and can be done by the primary clinicians seeing these patients (e.g. ENT surgeons, audiovestibular physicians and some paediatricians). Hence, ordering clinicians have to be aware of the requirements and management of results. Informed consenting is required, and NHSE-wide resources are being developed to support this. It is important to have a multidisciplinary team working, including genomic medicine input (e.g. in the Cambridge Genetic Hearing and Balance Loss Clinic, this is Dr Simon Holden), particularly to help with VUS’s, and for help with counselling for risks of transmission of the mutation, as well as non-auditory manifestations of syndromes that might be identified. It is useful to have both parents attend so blood can be taken for genetic testing, and a family tree proforma. Blood is banked for future genes that might be identified. It is helpful to try to phenotype the hearing loss, for help in future diagnostics of VUS’s. A common family number is assigned, so that members belonging to the same family can be identified. Lessons can be learned from other departments who may already have genetic disease clinics (e.g. ophthalmology, neurology). In some areas, this structure may form a dedicated MDT clinic, and in others it may consist of well-coordinated pathways for referral.
"Several commercial companies (e.g. Decibel Therapeutics, Akousos, Sensorion) are preparing human trials for otoferlin‑linked hearing loss, delivered intracochlearly"
Future gene therapies for hearing loss
Many gene therapy trials are pending implementation and there is rapid progress on this front. A very brief summary of approaches are:
Gene replacement therapies - delivery of a ‘trans gene’ that replaces the defective gene, commonly for recessive hearing loss, causing ‘loss of function’ mutations. The commonest considered vector for gene transfection is the adeno-associated virus (AAV). Several commercial companies (e.g. Decibel Therapeutics, Akouos, Sensorion) are preparing human trials for otoferlin-linked hearing loss, delivered intracochlearly. Many companies have other genes also in the pipeline, such as GJB2.
Gene suppression therapies - RNA-based therapies such as antisense oligonucleotides (ASOs), short interfering RNAs (siRNAs) and microRNAs (miRNAs) in dominantly inherited progressive hearing losses, in which there is interference with the normal gene by the protein from the defective allele. These therapies aim to silence the mRNA coding this abnormal interfering protein. Examples for potential use are in some forms of Ushers (USH1C).
CRISPR/Cas-9 Genome editing - this extremely powerful approach aims to directly edit a specific region of the defective gene. This promises to be particularly useful in dominant hearing loss, as many are due to single nucleotide substitutions. There are no current hearing loss trials in this space.
Auditory hair cell regeneration therapies - these therapies aim to regenerate hair cells from supporting cells. An example is the Hath 1 gene, and there is an ongoing trial in humans with this approach using adenovirus [3] with Novartis.
In addition to the above therapies, there are numerous drug-based therapies on the horizon that attempt to modify molecular pathways in hearing, and biohybrid approaches in which genes are combined with devices such as cochlear implants to improve neural health.
References
1. National Genomic Test Directory: Testing Criteria for Rare and Inherited Disease v3.1 August 2022.
www.england.nhs.uk/wp-content/
uploads/2018/08/Rare-and-inherited
-disease-eligibility-criteria-version
-3.1-August-2022.pdf
2. Ellard S, Baple EL, Callaway A, et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020.
www.acgs.uk.com/media/11631/
uk-practice-guidelines-for-variant
-classification-v4-01-2020.pdf
3. Novartis Pharmaceuticals. Safety, Tolerability and Efficacy for CGF166 in Patients With Unilateral or Bilateral Severe-to-profound Hearing Loss.
https://clinicaltrials.gov/ct2/
show/NCT02132130
All links last accessed July 2022.