Identifying the underlying genetic cause of hearing loss in newborns can improve dramatically the early diagnosis and appropriate intervention.
Hearing loss is the most common sensory disorder at birth, affecting approximately two out of 1000 newborns [1]. Congenital impaired hearing can be due to infections, ototoxic drugs, noise exposure, or have a genetic aetiology, with the latter being the cause of over than half of the cases [2]. The advent of whole exome sequencing (WES) has facilitated the discovery of deafness-related genes.
To date, pathogenic variants in more than 200 genes are associated with hearing loss in humans (http://deafnessvariationdatabase.org). Hearing loss has been shown to have major consequences on children, including speech and language development, academic performance and social skills [3]. Early detection of hearing loss has beneficial outcomes that were outlined in the World Health Organization 2010 report, including prompt medical management and rehabilitation (https://apps.who.int/iris/handle/10665/339288). Many countries have adopted such programmes in their healthcare system.
Newborn hearing is tested using automated auditory brainstem response (ABR), otoacoustic emissions (OAE), or both. These tests are part of routine healthcare in many countries, and are safe, quick, painless and inexpensive. These tests help detect disabling hearing loss as early as possible. Combining this screening with genetic diagnosis is predicted to significantly improve early detection, with implications for future care and habilitation.

Hereditary hearing loss is clinically and genetically heterogeneous and can be categorised into nonsyndromic and syndromic forms. Nonsyndromic hearing loss accounts for nearly 70% of all genetic hearing loss, with the majority of these cases inherited in an autosomal recessive pattern. The completion of the Human Genome Project in 2003 and the advent of next-generation sequencing have enabled rapid gene discovery and paved the way for personalised and precision medicine (Figure 1). In many developed countries, genomic sequencing is increasingly being performed for newborns who do not pass the traditional newborn hearing screen and have a negative CMV PCR test.
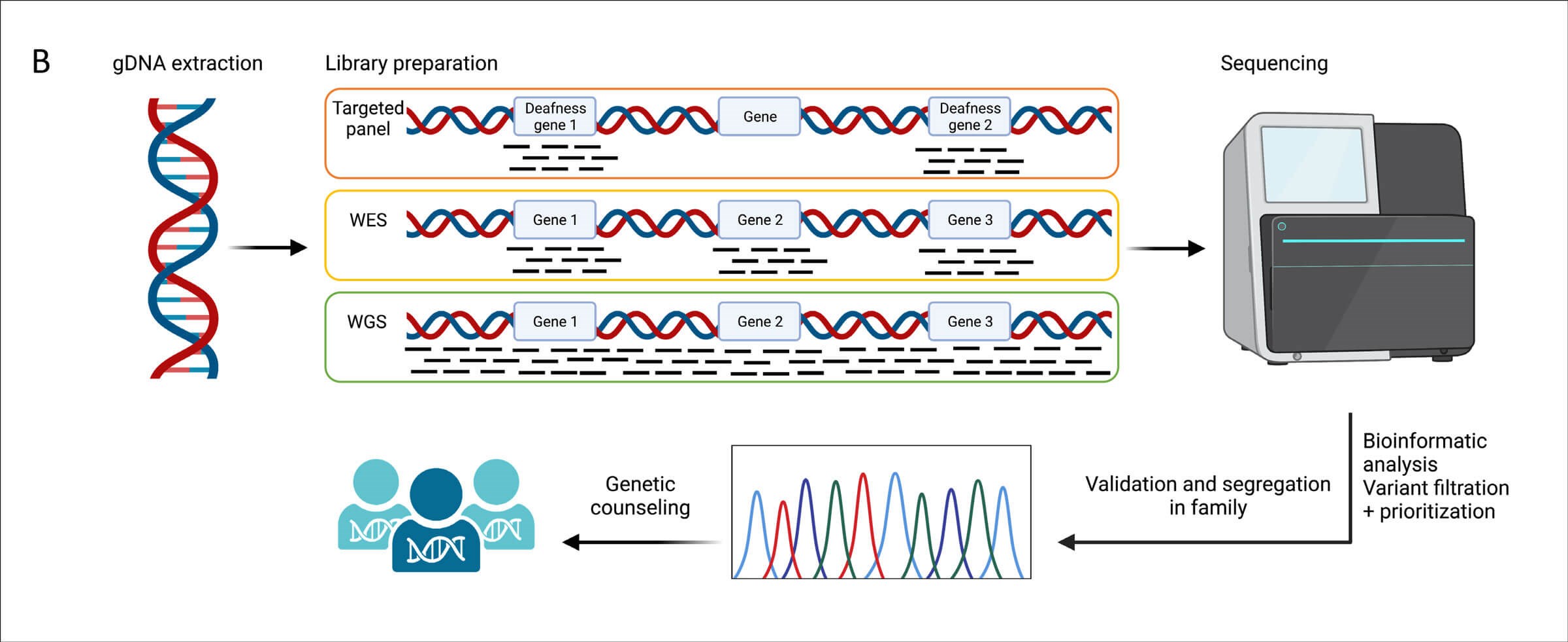
Figure 1. The pipeline of next generation sequencing-based genetic
testing in hearing loss diagnosis. Figure created with Biorender.
Genomic sequencing is expected to become the standard method of newborn screening in the next decade [4]. Different sequencing tests are available to identify the underlying genetic cause of hearing loss in humans (Figure 1). Whole genome sequencing (WGS) detects all variations in the human DNA (3x109 bp), while WES captures protein-coding genes (~20,000 genes) that encompasses 1-2% of the human genome. Hearing loss gene panels are also commonly used. There are multiple targeted gene panels designed to capture variations in hearing loss-related genes, spanning between 23-252 genes; and associated with syndromic and nonsyndromic forms of hearing loss and with multiple inheritance patterns (autosomal dominant, autosomal recessive, X-linked, and mitochondrial). Advantages and disadvantages of each test are outlined in Figure 2.
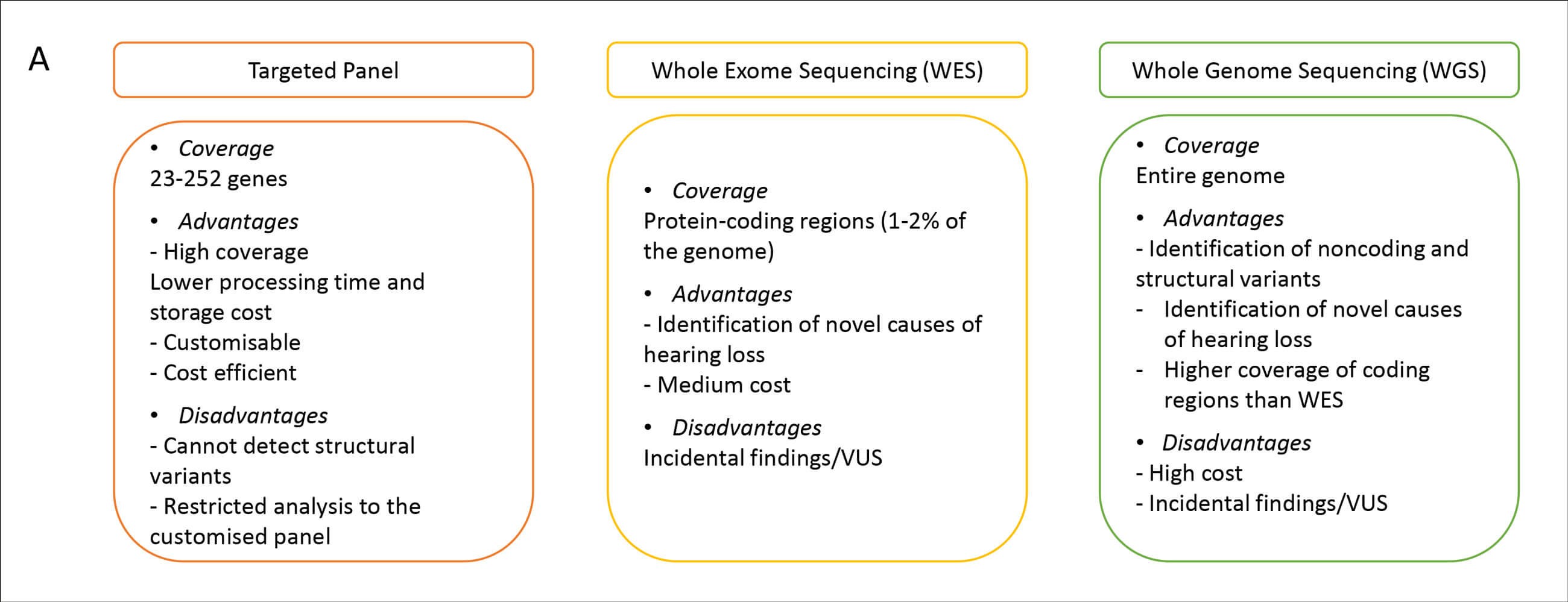
Figure 2. Comparison of targeted panels, exome and genome sequencing.
A comprehensive newborn screening approach consists of physiological, genetic and cCMV screening tests [5]. This approach will increase the number of newborns identified with hearing loss that were missed by the physiologic screen, and will enable faster intervention. Consequently, it will provide the opportunity for personalised genetic counselling, including risk assessment and prenatal testing by medical geneticists and genetic counsellors, in addition to improvements in medical management and rehabilitation by otolaryngologists and audiologists. In a recent study performed on a cohort of 8078 newborns in China, they assessed the combination of newborn hearing screen and genomic sequencing on the detection rate. There was a 15.6% increase in the diagnosis rate when genomic sequencing was integrated [6].
Integrating genomic sequencing into clinical settings provides an accurate diagnosis. Genetic diagnosis provides prognostic information on the severity and progression of hearing loss, and whether it is a non-syndromic or syndromic hearing loss. It may also provide parents with information regarding the chance of recurrence in future pregnancies. Genetic testing can also be important for the selection of the appropriate medical intervention and the likelihood of response to cochlear implantation. Up to 7% of cochlear implant (CI) recipients receive no benefit [7]. Several studies have analysed the association between genetic variants and the clinical outcome of cochlear implants. Children with GJB2 mutations, the most common cause of hereditary hearing loss, have shown an overall positive CI performance [8]. Similar favourable outcomes were seen in children with TMPRSS3 [9], LOXHD1 [10], MYO15A [11] and MYO6 [12]. However, patients with auditory neuropathy spectrum disorder have variable responses to CI depending on the genetic lesion site [13]. For example, TIMM8A, a spiral ganglion-expressed gene, is associated with poor CI performance, whereas OTOF, a membranous labyrinth-expressed gene, is associated with good outcomes post CI. Thus, combining newborn hearing screen and genetic testing can significantly enhance diagnosis, clinical management and prognosis. Given all the benefits of genetic testing for hearing loss, we must keep in mind that such testing raises a variety of social and ethical concerns, which include how the genetic data is used and the privacy of the patients, in addition to the social impact of genetic testing among the Deaf community [14].
In future, genetic testing may open the door to intervention by gene replacement using adeno-associated virus (AAV), gene modification by CRISPR/Cas9 or RNA modification by allele-specific oligonucleotides (ASO) [15]. Research on gene therapy to restore hearing loss is growing rapidly and has been successfully applied on mice models. These current achievements will aid the transition from proof-of-concept to clinical trials.
References
1. Korver AM, Smith RJH, Campet GV, et al. Congenital hearing loss. Nat Rev Dis Primers 2017;3:16094.
2. Lieu JEC, Kenna M, Anne A, Davidson L. Hearing loss in children: A review. JAMA 2020;324(21):2195-205.
3. Ciorba A, Bianchini C, Pelucchi S, Pastore A. The impact of hearing loss on the quality of life of elderly adults. Clin Interv Aging 2012;7:159-63.
4. Botkin JR, Rothwell E. Whole genome sequencing and newborn screening. Curr Genet Med Rep 2016;4(1):1-6.
5. Shearer AE, Shen J, Amret S, et al. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med 2019;21(11):2614-30.
6. Zhu Y, Hu L, Yang L, et al. Association between expanded genomic sequencing combined with hearing screening and detection of hearing loss among newborns in a neonatal intensive care unit. JAMA Netw Open 2022;5(7):e2220986.
7. Raine CH, Summerfield Q, Strachan DR, et al. The cost and analysis of nonuse of cochlear implants. Otol Neurotol 2008;29(2):221-4.
8. Yan YJ, Li Y, Yang T, et al. The effect of GJB2 and SLC26A4 gene mutations on rehabilitative outcomes in pediatric cochlear implant patients. Eur Arch Otorhinolaryngol 2013;270(11):2865-70.
9. Chen YS, Cabrera E, Tucker BJ, et al., TMPRSS3 expression is limited in spiral ganglion neurons: implication for successful cochlear implantation. J Med Genet 2022;59(12):1219-26
10. Maekawa K, Nishio SY, Abe S, et al. Mutational Spectrum and Clinical Features of Patients with LOXHD1 Variants Identified in an 8074 Hearing Loss Patient Cohort. Genes (Basel) 2019;10(10):735.
11. Liu WH, Chang PY, Changet SC, et al. Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS One 2019;14(1):e0211261.
12. Volk AE, Lang-Roth R, Yigit G, et al. A novel MYO6 splice site mutation causes autosomal dominant sensorineural hearing loss type DFNA22 with a favourable outcome after cochlear implantation. Audiol Neurootol 2013;18(3):192-9.
13. Shearer AE, Hansen MR. Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Investig Otolaryngol 2019;4(4):429-40.
14. Burton SK, Withrow K, Arnoset KS, et al. A focus group study of consumer attitudes toward genetic testing and newborn screening for deafness. Genet Med 2006;8(12):779-83.
15. Taiber S, Avraham KB. Genetic therapies for hearing loss: Accomplishments and remaining challenges. Neurosci Lett 2019;713:134527.