Whilst snoring and obstructive sleep apnoea are relatively common diagnoses in paediatric ENT, children with craniofacial syndromes take the problem to the next level. Robert Nash and Michelle Wyatt describe the Great Ormond Street multidisciplinary approach to treating this complex group.
Craniofacial syndromes comprise of a group of rare conditions leading to significant dysmorphism of the cranium, maxilla and mandible. Broadly speaking, these syndromes may be divided into those characterised by either craniosynostosis, or clefts.
The term craniosynostosis refers to a group of conditions causing the premature fusion of the cranial sutures, with consequent effects on the development of the skull, face and brain. This can lead to raised intracranial pressure, subluxation of the globe and upper airway obstruction. In syndromic craniosynostosis, this process is associated with fibroblast growth factor receptor (FGFR) gene mutations [1].
Craniofacial syndromes associated with cleft comprise a more heterogenous group, in which mandibular hypoplasia is commonly an important feature. The most notable of these syndromes is Pierre Robin sequence, although numerous other syndromes including Treacher Collins, Nagar and Stickler syndromes are also included.
Otolaryngologists may see patients with these syndromes for a variety of reasons. These groups have a high incidence of hearing loss, distressing rhinological symptoms and speech delay. Most notably however, upper airway obstruction is common and may be severe. It is not uncommon for these patients to be at the most severe end of a spectrum ranging from ‘simple snoring’, through obstructive sleep apnoea (OSA) and on to daytime stertor and desaturation.
The anatomical nature of this upper airway obstruction varies between each syndrome. Broadly speaking, however, syndromic craniosynostosis is associated with maxillary, and consequently midface, hypoplasia. This worsens the nasal airway and leads to a restricted and crowded pharynx [2]. In cleft syndromes, mandibular hypoplasia with glossoptosis may lead to obstruction at the level of the tongue base [3].
The prevalence of upper airway obstruction is sufficient for screening sleep studies to be performed on patients referred to craniofacial teams. The incidence of OSA in syndromic craniosynostosis is about 50%, and in Pierre Robin sequence may be around 80%. Overall, the odds ratio for a diagnosis of OSA in children with any craniofacial anomaly is 38 [4]. A number of patients will already have airway adjuncts in place to manage the airway prior to referral to specialist teams. This is indicative of the severity of obstruction that may be constantly symptomatic, rather than only manifesting itself during sleep.
“Management of sleep disordered breathing in children with craniofacial syndromes is a multidisciplinary process in which neurological, ophthalmological and airway compromise has to be considered.”
When screening this patient group, formal polysomnography is preferred to pulse oximetry. There are a number of reasons for this. Most importantly, central sleep apnoea may be a confounding factor. This is particularly relevant in patients with craniosynostosis and consequent raised intracranial pressure. Other neurological comorbidities are more common in these patients and may confound the findings of oximetry. Furthermore, it is recognised that these patients may present with ‘respiratory effort related arousals’ (RERAs) and consequent sleep disordered breathing (SDB) even in the absence of desaturation.
Transcutaneous carbon dioxide monitoring can help assess the degree of obstruction and the effect it has on the patient’s physiology. It is not uncommon for sleep studies to be repeated. This may be indicated by a disparity with clinical impression, unrepresentative sleeping conditions or to assess response to airway adjuncts. Broadly speaking, it is useful for clinicians to have as much data available as possible when making treatment decisions that may have significant ramifications for the patient and their family.
The management of OSA in these groups of patients is highly challenging, and is best conducted in a multidisciplinary setting. The methods used to treat OSA have significant overlap with methods used to treat adult and paediatric OSA, although some techniques specific to these patients are also used.
In common with general paediatric practice for the management of SDB, there is a role for adenotonsillectomy in this patient group. It is, however, clear that adenotonsillectomy is much less effective at relieving SDB in patients with craniofacial syndromes. This is particularly true for children with ‘cleft syndromes’ such as Pierre Robin syndrome. Adenotonsillectomy has limited efficacy in improving the airway at the site of the tongue base. Furthermore, in patients with cleft palate, adenoidectomy may be relatively contraindicated.
In these situations, it is not uncommon for a partial adenoidectomy to be considered, dependent on the severity of OSA, the degree of adenotonsillar hypertrophy and speech development. Adenotonsillectomy is more commonly used in syndromic craniosynostosis, and it is credited with a reduction in the need for tracheostomy [5]. However, the evidence is mixed, and it is therefore not undertaken in every patient with SDB, but rather is an option for consideration during a multidisciplinary assessment.
Continuous positive airway pressure (CPAP) devices are also used in these patients. A need for relatively high airway pressures, a paediatric patient group and suboptimal treatment efficacy make compliance with treatment challenging. However, CPAP may be a safe and reliable option that may allow surgical management to be deferred.
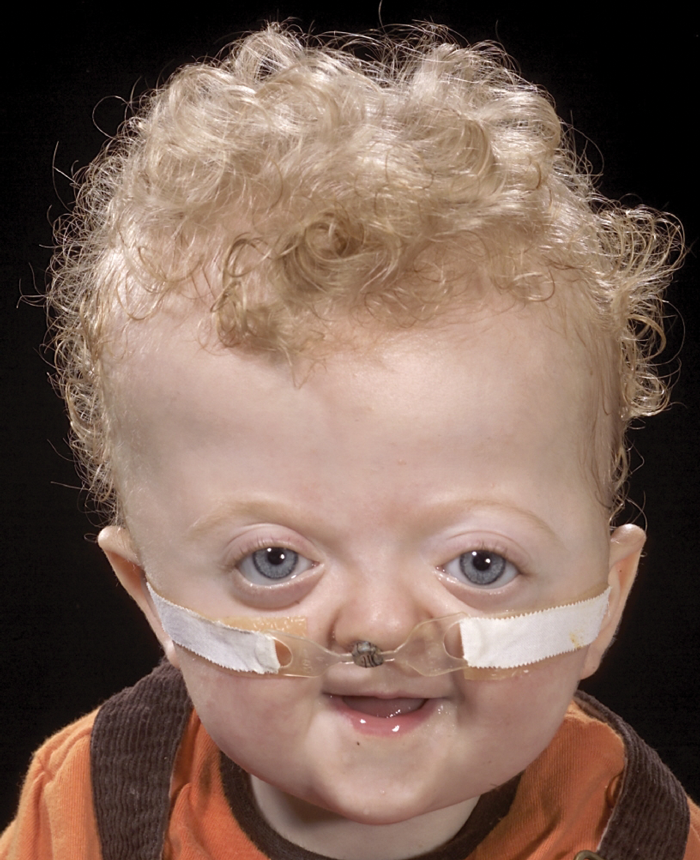
Figure 1: A nasal prong in place in a child with syndromic craniosynostosis.
Another strategy that may defer definitive management of SDB is the use of nasopharyngeal airways or ‘nasal prongs’, see Figure 1. These may be placed to bypass nasal and nasopharyngeal obstruction, and their presence may ‘stent’ oropharyngeal obstruction. As such these may be used in patients with Pierre Robin [6] and craniosynostosis.
Tracheostomy is considered the definitive management strategy for OSA in all patient groups. However, in a general adult or paediatric population it is only very rarely undertaken solely for this indication. The exception to this is in children with craniofacial syndromes, where tracheostomy is considered much more frequently. This is largely due to the severity of the upper airway obstruction, although concomitant glottic pathology and planned craniofacial surgery may also predispose to tracheostomy. However, tracheostomy is not without risk, and maintenance of a tracheostomy in a child is difficult and significantly impairs quality of life for the child and family. It is a recognition of these problems that has led increasingly to the use of other management strategies.
Craniofacial surgery is undertaken for a variety of reasons in these patients. Cosmesis, raised intracranial pressure and proptosis may all be addressed with these procedures, particularly in patients with craniosynostosis. The airway is also an important consideration, and the timing of this surgery does take into account the severity of the upper airway obstruction. For a proportion of patients these procedures are effective in providing an improved airway [7,8], however, it can be difficult to predict which patients will benefit and which will continue to have airway obstruction. These operations are major undertakings, with recognised morbidity, and intensive postoperative care is required. If performed at an early age, revision may be required at a later date as the child grows.
In summary, management of sleep disordered breathing in children with craniofacial syndromes is a multidisciplinary process in which neurological, ophthalmological and airway compromise has to be considered. There is an important role for the otolaryngologist, respiratory physician, craniofacial surgeon and paediatrician in balancing the appropriate interventions with the manifestation of these challenging pathologies.
References
1. Agochukwu NB, Solomon BD, Muenke M. Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses. Childs Nerv Syst 2012;28(9):1447-63.
2. de Water VR, Saridin JK, Bouw F, Murawska MM, Koudstaal MJ. Measuring upper airway volume: accuracy and reliability of Dolphin 3D software compared to manual segmentation in craniosynostosis patients. J Oral Maxillofac Surg 2014;72(1):139-44.
3. Salerno S, Gagliardo C, Vitabile S, Militello C, La Tona G, Giuffrè M, et al. Semi-automatic volumetric segmentation of the upper airways in patients with Pierre Robin sequence. Neuroradiol J 2014;27(4):487-94.
4. Lam DJ, Jensen CC, Mueller BA, Starr JR, Cunningham ML, Weaver EM. Pediatric sleep apnea and craniofacial anomalies: a population-based case-control study. Laryngoscope 2010;120(10):2098-105.
5. Amonoo-Kuofi K, Phillips SP, Randhawa PS, Lane R, Wyatt ME, Leighton SE. Adenotonsillectomy for sleep-disordered breathing in children with syndromic craniosynostosis. J Craniofac Surg 2009;20(6):1978-80.
6. Abel F, Bajaj Y, Wyatt M, Wallis C. The successful use of the nasopharyngeal airway in Pierre Robin sequence: an 11-year experience. Arch Dis Child 2012;97(4):331-4.
7. Tahiri Y, Viezel-Mathieu A, Aldekhayel S, Lee J, Gilardino M. The effectiveness of mandibular distraction in improving airway obstruction in the pediatric population. Plast Reconstr Surg 2014;133(3):352e-359e.
8. Mathijssen I, Arnaud E, Marchac D, Mireau E, Morisseau-Durand MP, Guérin P, Renier D. Respiratory outcome of mid-face advancement with distraction: a comparison between Le Fort III and frontofacial monobloc. J Craniofac Surg 2006;17(5):880-2.
Declaration of competing interests: None declared.



