Shahar Taiber and Karen Avraham give us a summary of gene therapies for hearing loss, with an overview of limitations and what the future holds.
Hearing loss is the most common sensory disorder. The last two decades have seen a revolution in the discovery of genes involved in hearing loss and cellular pathways implicated in age, noise, and drug-induced hearing loss. Advances in gene discovery and basic research have created opportunities to try to treat or cure deafness using gene therapy.
Gene therapy in deafness is now coming of age, with an increasing number of studies reporting successful rescue of auditory function in animal models of genetic deafness, noise-induced hearing loss (NIHL), and age-related hearing loss (ARHL).
Hearing loss can result from both genetic variants and environmental factors, such as noise exposure, ageing, certain drugs, and more. It is estimated that over half of the cases of congenital deafness are caused by genetic variants [1]. On the other side of the spectrum, over half of the population over 75-years-old is impacted by ARHL, which is associated with multiple comorbidities and is considered a major risk factor for dementia. Genetic hearing loss can be inherited in a dominant, recessive or X-linked mode, and through mutations in mitochondria inherited though the maternal line. Genetic variants may encompass large chromosomal regions, single base pair changes, or a variety of alterations that are in between these two extremes. Regulatory variants, in the non-coding regions of the genome, as also associated with deafness, though knowledge of these is very scarce.
“Previous studies demonstrated successful gene silencing using gene editing technologies where a mutant allele was deactivated, rather than corrected”
Over 110 genes are associated with hearing loss [1]. These genes encode a large variety of proteins, each with essential functions in the auditory system. Many of these genes are transcription factors involved in the development of the auditory system, orchestrating unique transcriptional programmes in specific cell types. An example of this is the gene POU4F3, initially identified for progressive hearing loss in an extended Israeli family [2], which is involved in the regulation of hair-cell genes. Another group of genes includes transporters and channels, which are for maintaining homeostasis by influencing ion concentrations, pH levels, and cellular and compartment potentials. A prominent member of this group is the gene GJB2, encoding the protein connexin 26. This form of genetic deafness is the most common one, responsible for 30-50% of genetic deafness worldwide [3]. Another critical group of genes is associated with intra and extracellular structural proteins, such as MYO6, encoding a non-conventional myosin expressed by hair cells [4], and TECTA, encoding an extracellular component of the tectorial membrane [5].
RELATED TOPIC - Tap the box below

Various methods are being developed to treat the genetic forms of deafness described above, including gene replacement, gene silencing, gene editing, and splicing modulation. Gene replacement involves delivering a wild-type copy of a mutated gene in-trans alongside regulatory elements, typically delivered by replication-deficient recombinant adeno-associated virus (AAV) vectors. This approach is mostly relevant in cases of recessive deafness and autosomal deafness stemming from haploinsufficiency. This form of therapy has been successfully applied in mouse models for human deafness for TMC1, USH1C, WHRN, OTOF, SYNE4, and more [6,7].
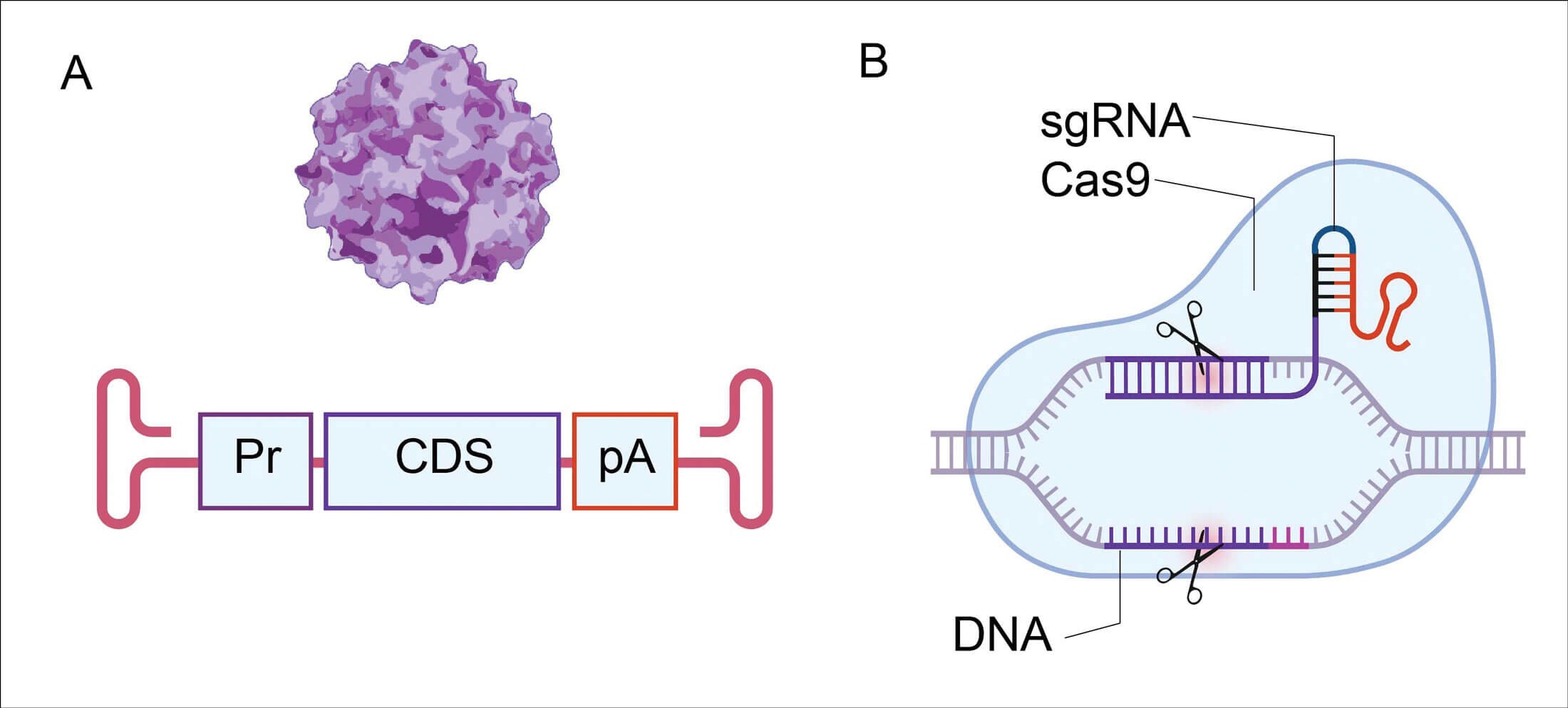
Approaches used in gene therapy for hearing loss.
A: Adeno-associated virus (AAV) vector containing a gene promoter (Pr),
coding sequence (CDS), and a poly-adenylation signal (pA).
B: Scheme of CRISPR/Cas9, with scissors demonstrating editing sites.
Figure created with Biorender.
Gene editing technologies, predominantly those based on CRISPR/Cas9, have long been anticipated to be applied to correct mutations. Recently, this has been achieved for the first time in a mouse model of TMC1 deafness, where a deactivated Cas9 protein fused to a cytosine deaminase, a system named ‘base editing’, was used to restore wild-type TMC1 expression and alleviate hearing loss [8]. Previous studies demonstrated successful gene silencing using gene editing technologies where a mutant allele was deactivated, rather than corrected [9]. This approach is relevant for some forms of dominant deafness, where the mutation creates a toxic product. Other methods to mediate silencing include small-interfering RNA (siRNA) and antisense oligonucleotides (ASO). ASO can also be used to correct splicing, as was achieved in mouse models of USH1C deafness [10].
“While AAV vectors are considered to be relatively safe and have shown robust transduction of cell types in the inner ear of mice and non-human primates, hurdles remain”
Of note, gene editing approaches need to be tailored for specific mutations, not just genes, whereas gene replacement potentially covers different mutations in the same gene. However, since gene expression is regulated by various complex mechanisms, the endogenous genomic context can be optimal in this sense, as it circumvents the need for regulatory elements, such as promoters and poly-adenylation sequences.
Delivery approaches are a key factor in implementing gene therapy. While AAV vectors are considered to be relatively safe and have shown robust transduction of cell types in the inner ear of mice and non-human primates, hurdles remain [7]. High doses of AAV vectors can be lethal in humans and primates, and the long-term safety of low doses delivered to the inner ear need to be characterised. Immune responses against viral capsid proteins after inner ear injection have been reported, which could potentially also impede therapeutic efficacy. Moreover, the 4.7 kb capacity limit of AAV hinders delivery of large genes. This limitation can be bypassed by splitting the transgene into two vectors. Finally, scaling and production of AAV vectors in large quantities is required for translating this therapy to humans.
“Gene therapy holds great promises for the field of hearing loss and has already shown encouraging preclinical achievements”
To summarise, gene therapy holds great promises for the field of hearing loss and has already shown encouraging preclinical achievements. A growing number of companies are showing interest in this field and clinical trials might be around the corner, with one already behind us. It will be crucial to move forward with caution and to address the needs of the community as a whole, putting efforts into rare forms of deafness as well, and making these treatments as accessible as possible in all communities and countries around the world.
References
1. Sheffield AM, Smith RJH. The epidemiology of deafness. Cold Spring Harb Perspect Med 2019;9(9):a033258.
2. Vahava O, Morell R, Lynch ED, et al. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 1998;279(5358):1950-4.
3. Nickel R, Forge A. Gap junctions and connexins in the inner ear: their roles in homeostasis and deafness. Curr Opin Otolaryngol Head Neck Surg 2008;16(5):452‑7.
4. Avraham KB, Hasson T, Steel KP, et al. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nature Genetics 1995;11(4):369-75.
5. Verhoeven K, Van Laer L, Kirschhofer K, et al. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nature Genetics 1998;19(1):60-2.
6. Taiber S, Cohen R, Yizhar-Barnea O, et al. Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol Med 2021;13(2):e13259.
7. Delmaghani S, El-Amraoui A. Inner ear gene therapies take off: Current promises and future challenges. J Clin Med 2020;9(7):2309.
8. Yeh WH, Shubina-Oleinik O, Levy JM, et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci Transl Med 2020;12(546):eaay9101.
9. Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018;553(7687):217-21.
10. Wang L, Kempton JB, Jiang H, et al. Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction. Nucleic Acids Res 2020;48(9):5065 80.
RELATED TOPIC - Tap the box below





